Introdução
O que você precisa saber de cara
Visão geral
A catarata das suturas de início precoce é uma condição rara que afeta a transparência do cristalino (a lente natural do olho) desde os primeiros dias ou meses de vida. Nessa condição, a opacidade se forma especificamente nas chamadas 'suturas' do cristalino, que são as linhas de junção das fibras que compõem essa estrutura. A doença pode estar presente ao nascimento (neonatal) ou surgir ainda na primeira infância.[1]
Sinais e sintomas
Os principais sinais da catarata das suturas de início precoce são a opacidade visível no cristalino e a consequente redução da visão. Como a doença se manifesta muito cedo, os bebês podem apresentar dificuldade para fixar o olhar, movimentos oculares anormais (nistagmo) ou sensibilidade excessiva à luz (fotofobia). A gravidade da perda visual varia conforme a extensão e a localização da opacidade.[1]
Causas genéticas
A catarata das suturas de início precoce é causada por alterações (mutações) em genes que produzem proteínas essenciais para a transparência e a estrutura do cristalino. Os genes já identificados incluem: CRYGS (cristalina gama S), MIP (proteína intrínseca maior das fibras do cristalino), CRYBB2 (cristalina beta B2), CRYBA1 (cristalina beta A3), BFSP2 (faquinina) e GJA8 (proteína alfa-8 das junções comunicantes). Cada um desses genes fornece instruções para a produção de proteínas que mantêm o cristalino transparente e organizado. Mutações nesses genes podem interromper esse processo, levando à formação da catarata.[1][3]
Diagnóstico
O diagnóstico é realizado por um oftalmologista, que examina o olho com instrumentos especiais (lâmpada de fenda) para visualizar a opacidade característica nas suturas do cristalino. Para confirmar a causa genética, podem ser solicitados exames como o cariótipo (análise dos cromossomos), a pesquisa de microdeleções/microduplicações por FISH, o sequenciamento completo do exoma (WES) e a dosagem de alfa-fetoproteína. Atualmente, existem 336 testes genéticos disponíveis para essa condição, e 134 variantes genéticas já foram registradas no banco de dados ClinVar.[1][3]
Tratamento e manejo
O tratamento da catarata das suturas de início precoce é essencialmente cirúrgico, com a remoção do cristalino opaco (facoemulsificação ou cirurgia de catarata). Em bebês e crianças, a cirurgia deve ser realizada o mais cedo possível para evitar a ambliopia (olho preguiçoso) e permitir o desenvolvimento visual adequado. Após a cirurgia, é necessário o uso de lentes de contato ou óculos para corrigir a visão. O acompanhamento com oftalmologista e equipe multidisciplinar é fundamental. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para essa condição, incluindo atendimento em reabilitação para doenças raras.[1]
Prognóstico e qualidade de vida
O prognóstico visual depende da precocidade do diagnóstico e do tratamento. Quando a cirurgia é realizada nos primeiros meses de vida e o acompanhamento pós-operatório é adequado, muitas crianças conseguem desenvolver visão funcional. No entanto, podem persistir desafios como a necessidade de usar óculos ou lentes de contato por toda a vida e o risco de complicações como glaucoma ou descolamento de retina. O suporte de uma equipe multidisciplinar (oftalmologista, geneticista, psicólogo, terapeuta ocupacional) é essencial para maximizar a qualidade de vida e a inclusão social.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Catarata hereditária de início precoce, afetando o cristalino. Associada a mutações nos genes CRYGS, MIP, CRYBB2, CRYBA1 e BFSP2. Manifesta-se como opacidades suturais no cristalino, prejudicando a visão.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A catarata das suturas de início precoce é uma condição rara que afeta a transparência do cristalino (a lente natural do olho) desde os primeiros dias ou meses de vida. Nessa condição, a opacidade se forma especificamente nas chamadas 'suturas' do cristalino, que são as linhas de junção das fibras que compõem essa estrutura. A doença pode estar presente ao nascimento (neonatal) ou surgir ainda na primeira infância.[1]
Sinais e sintomas
Os principais sinais da catarata das suturas de início precoce são a opacidade visível no cristalino e a consequente redução da visão. Como a doença se manifesta muito cedo, os bebês podem apresentar dificuldade para fixar o olhar, movimentos oculares anormais (nistagmo) ou sensibilidade excessiva à luz (fotofobia). A gravidade da perda visual varia conforme a extensão e a localização da opacidade.[1]
Causas genéticas
A catarata das suturas de início precoce é causada por alterações (mutações) em genes que produzem proteínas essenciais para a transparência e a estrutura do cristalino. Os genes já identificados incluem: CRYGS (cristalina gama S), MIP (proteína intrínseca maior das fibras do cristalino), CRYBB2 (cristalina beta B2), CRYBA1 (cristalina beta A3), BFSP2 (faquinina) e GJA8 (proteína alfa-8 das junções comunicantes). Cada um desses genes fornece instruções para a produção de proteínas que mantêm o cristalino transparente e organizado. Mutações nesses genes podem interromper esse processo, levando à formação da catarata.[1][3]
Diagnóstico
O diagnóstico é realizado por um oftalmologista, que examina o olho com instrumentos especiais (lâmpada de fenda) para visualizar a opacidade característica nas suturas do cristalino. Para confirmar a causa genética, podem ser solicitados exames como o cariótipo (análise dos cromossomos), a pesquisa de microdeleções/microduplicações por FISH, o sequenciamento completo do exoma (WES) e a dosagem de alfa-fetoproteína. Atualmente, existem 336 testes genéticos disponíveis para essa condição, e 134 variantes genéticas já foram registradas no banco de dados ClinVar.[1][3]
Tratamento e manejo
O tratamento da catarata das suturas de início precoce é essencialmente cirúrgico, com a remoção do cristalino opaco (facoemulsificação ou cirurgia de catarata). Em bebês e crianças, a cirurgia deve ser realizada o mais cedo possível para evitar a ambliopia (olho preguiçoso) e permitir o desenvolvimento visual adequado. Após a cirurgia, é necessário o uso de lentes de contato ou óculos para corrigir a visão. O acompanhamento com oftalmologista e equipe multidisciplinar é fundamental. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para essa condição, incluindo atendimento em reabilitação para doenças raras.[1]
Prognóstico e qualidade de vida
O prognóstico visual depende da precocidade do diagnóstico e do tratamento. Quando a cirurgia é realizada nos primeiros meses de vida e o acompanhamento pós-operatório é adequado, muitas crianças conseguem desenvolver visão funcional. No entanto, podem persistir desafios como a necessidade de usar óculos ou lentes de contato por toda a vida e o risco de complicações como glaucoma ou descolamento de retina. O suporte de uma equipe multidisciplinar (oftalmologista, geneticista, psicólogo, terapeuta ocupacional) é essencial para maximizar a qualidade de vida e a inclusão social.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
6 genes identificados com associação a esta condição.
Crystallins are the dominant structural components of the vertebrate eye lens
Cataract 20, multiple types
An opacification of the crystalline lens of the eye that frequently results in visual impairment or blindness. Opacities vary in morphology, are often confined to a portion of the lens, and may be static or progressive. In general, the more posteriorly located and dense an opacity, the greater the impact on visual function. CTRCT20 includes progressive polymorphic anterior, posterior, or peripheral cortical.
Aquaporins form homotetrameric transmembrane channels, with each monomer independently mediating water transport across the plasma membrane along its osmotic gradient (PubMed:11001937, PubMed:24120416). Specifically expressed in lens fiber cells, this aquaporin is crucial for maintaining lens water homeostasis and transparency. Beyond water permeability, it also acts as a cell-to-cell adhesion molecule, forming thin junctions between lens fiber cells that are essential for maintaining the ordere
Cell membraneCell junction
Cataract 15, multiple types
An opacification of the crystalline lens of the eye that frequently results in visual impairment or blindness. Opacities vary in morphology, are often confined to a portion of the lens, and may be static or progressive. CTRCT15 includes polymorphic, progressive punctate lamellar, cortical, anterior and posterior polar, nonprogressive lamellar with sutural opacities, embryonic nuclear, and pulverulent cortical, among others.
Crystallins are the dominant structural components of the vertebrate eye lens
Cataract 3, multiple types
An opacification of the crystalline lens of the eye that frequently results in visual impairment or blindness. Opacities vary in morphology, are often confined to a portion of the lens, and may be static or progressive. CTRCT3 includes congenital cerulean and sutural cataract with punctate and cerulean opacities, among others. Cerulean cataract is characterized by peripheral bluish and white opacifications organized in concentric layers with occasional central lesions arranged radially. The opacities are observed in the superficial layers of the fetal nucleus as well as the adult nucleus of the lens. Involvement is usually bilateral. Visual acuity is only mildly reduced in childhood. In adulthood, the opacifications may progress, making lens extraction necessary. Histologically the lesions are described as fusiform cavities between lens fibers which contain a deeply staining granular material. Although the lesions may take on various colors, a dull blue is the most common appearance and is responsible for the designation cerulean cataract. Sutural cataract with punctate and cerulean opacities is characterized by white opacification around the anterior and posterior Y sutures, and grayish and bluish, spindle shaped, oval punctate and cerulean opacities of various sizes arranged in lamellar form. The spots are more concentrated towards the peripheral layers and do not delineate the embryonal or fetal nucleus. Phenotypic variation with respect to the size and density of the sutural opacities as well as the number and position of punctate and cerulean spots is observed among affected subjects.
Crystallins are the dominant structural components of the vertebrate eye lens
Cataract 10, multiple types
An opacification of the crystalline lens of the eye that frequently results in visual impairment or blindness. Opacities vary in morphology, are often confined to a portion of the lens, and may be static or progressive. CTRCT10 includes congenital zonular with sutural opacities, among others. This is a form of zonular cataract with an erect Y-shaped anterior and an inverted Y-shaped posterior sutural opacities. Zonular or lamellar cataracts are opacities, broad or narrow, usually consisting of powdery white dots affecting only certain layers or zones between the cortex and nucleus of an otherwise clear lens. The opacity may be so dense as to render the entire central region of the lens completely opaque, or so translucent that vision is hardly if at all impeded. Zonular cataracts generally do not involve the embryonic nucleus, though sometimes they involve the fetal nucleus. Usually sharply separated from a clear cortex outside them, they may have projections from their outer edges known as riders or spokes.
Required for the correct formation of lens intermediate filaments as part of a complex composed of BFSP1, BFSP2 and CRYAA (PubMed:28935373). Plays a role in maintenance of retinal lens optical clarity (By similarity)
Cell membraneCytoplasmCytoplasm, cytoskeletonCytoplasm, cell cortex
Cataract 12, multiple types
An opacification of the crystalline lens of the eye that frequently results in visual impairment or blindness. Opacities vary in morphology, are often confined to a portion of the lens, and may be static or progressive. In general, the more posteriorly located and dense an opacity, the greater the impact on visual function. The opacities can be nuclear, sutural, stellate cortical, lamellar, cortical, nuclear embryonic, Y-sutural, punctate cortical, congenital or with juvenile- and adult-onset.
Structural component of eye lens gap junctions (PubMed:18006672, PubMed:19756179). Gap junctions are dodecameric channels that connect the cytoplasm of adjoining cells. They are formed by the docking of two hexameric hemichannels, one from each cell membrane (By similarity). Small molecules and ions diffuse from one cell to a neighboring cell via the central pore (PubMed:18006672, PubMed:19756179)
Cell membraneCell junction, gap junction
Cataract 1, multiple types
An opacification of the crystalline lens of the eye that frequently results in visual impairment or blindness. Opacities vary in morphology, are often confined to a portion of the lens, and may be static or progressive. CTRCT1 includes congenital, zonular pulverulent, nuclear progressive, nuclear pulverulent, nuclear total, total, and posterior subcapsular types of cataract. Zonular or lamellar cataracts are opacities, broad or narrow, usually consisting of powdery white dots affecting only certain layers or zones between the cortex and nucleus of an otherwise clear lens. The opacity may be so dense as to render the entire central region of the lens completely opaque, or so translucent that vision is hardly if at all impeded. Zonular cataracts generally do not involve the embryonic nucleus, though sometimes they involve the fetal nucleus. Usually sharply separated from a clear cortex outside them, they may have projections from their outer edges known as riders or spokes. In some cases cataract is associated with microcornea without any other systemic anomaly or dysmorphism. Microcornea is defined by a corneal diameter inferior to 10 mm in both meridians in an otherwise normal eye.
Variantes genéticas (ClinVar)
134 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Catarata das suturas de início precoce
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
A novel ADAMTSL4 compound heterozygous mutation in isolated ectopia lentis: a case report and review of the literature.
Congenital ectopia lentis is characterized by dislocation of the lens caused by partial or complete abnormalities in the zonular fibers. It can be caused by either systemic diseases or isolated ocular diseases. Gene detection techniques can provide valuable information when an etiological diagnosis is challenging. Herein, we report the case of a six-year-old girl with a confirmed diagnosis of isolated ectopia lentis caused by a compound heterozygous ADAMTSL4 gene mutation. The patient was a 6-year-old Chinese Han girl with strabismus in the right eye. Slit lamp examination revealed that the lens in the right eye was opacified and dislocated, without an ectopic pupil. Gene detection demonstrated the presence of a compound heterozygous mutation in the ADAMTSL4 gene [c. 2270dupG (p.Gly758Trpfs *59) and c. 2110A > G (p.Ser704Gly)], and the diagnosis of isolated ectopia lentis was confirmed. She underwent lens extraction, and a sutured scleral-fixated posterior chamber intraocular lens (IOL) was placed in the right eye. The best-corrected visual acuity was 0.1 one month postoperatively. Gene detection plays a crucial role in diagnosing disorders with similar symptoms, such as isolated ectopia lentis and Marfan syndrome. In this study, we used whole exons sequencing to diagnose isolated ectopia lentis and identified the variant c.2110A > G (p.Ser704Gly), which may be associated with the development of ectopia lentis and early-onset cataract in the patient. These pathogenic gene mutations have significant implications for the genetic diagnosis of congenital ectopia lentis, treatment, surveillance, and hereditary and prenatal counseling for the patient and their family members.
Hereditary cataract in the Bengal cat in Poland.
This paper reports the significant prevalence of a presumed hereditary cataract in the Bengal cat breed in Poland. The nuclear part of the lens is affected and previous reports from Sweden and France for this type of feline cataract suggest that a recessive mode of inheritance is probably involved. Presumed congenital or neonatal cataract involving the posterior nuclear part of each lens was initially diagnosed in a 12 month old male Bengal cat. As both parents and a sibling were also affected with cataract, a group of 18 related and 11 non-related cats was then subsequently examined. Eight related cats and one non-related cat were found to be similarly affected. A breed survey was then completed using an additional five centres across Poland and a further 190 related cats were examined. A total of 223 cats have been involved in this study, with 75 (33%) being affected with several types of cataract and 67 (30%) being specifically affected with the same or similar nuclear lesions. Eight cats (3.6%) presented with other cataract types and a prominence of the posterior lens suture lines was recorded in 65 cats unaffected with cataract (29%). There were no demonstrable vision problems. Neither age nor coat colour was significantly associated with the nuclear cataract, but the nuclear cataract group had a higher proportion of females than the unaffected group. Pedigree analysis has indicated probable inheritance as a recessive trait. These findings suggest that a presumably inherited nuclear cataract is present in the Bengal cat breed in Poland. It is considered to be either congenital or of very early onset, probably being inherited as a recessive trait. Although the lesion has no noticeable effect on vision, breeders in Poland and worldwide should be aware of the disease and clinical examination of young breeding stock prior to reproduction is advisable.
Human βA3/A1-crystallin splicing mutation causes cataracts by activating the unfolded protein response and inducing apoptosis in differentiating lens fiber cells.
βγ-Crystallins, having a uniquely stable two domain four Greek key structure, are crucial for transparency of the eye lens,. Mutations in lens crystallins have been proposed to cause cataract formation by a variety of mechanisms most of which involve destabilization of the protein fold. The underlying molecular mechanism for autosomal dominant zonular cataracts with sutural opacities in an Indian family caused by a c.215+1G>A splice mutation in the βA3/A1-crystallin gene CRYBA1 was elucidated using three transgenic mice models. This mutation causes a splice defect in which the mutant mRNA escapes nonsense mediated decay by skipping both exons 3 and 4. Skipping these exons results in an in-frame deletion of the mRNA and synthesis of an unstable p.Ile33_Ala119del mutant βA3/A1-crystallin protein. Transgenic expression of mutant βA3/A1-crystallin but not the wild type protein results in toxicity and abnormalities in the maturation and orientation of differentiating lens fibers in c.97_357del CRYBA1 transgenic mice, leading to a small spherical lens, cataract, and often lens capsule rupture. On a cellular level, the lenses accumulated p.Ile33_Ala119del βA3/A1-crystallin with resultant activation of the stress signaling pathway - unfolded protein response (UPR) and inhibition of normal protein synthesis, culminating in apoptosis. This highlights the mechanistic contrast between mild mutations that destabilize crystallins and other proteins, resulting in their being bound by the α-crystallins that buffer lens cells against damage by denatured proteins, and severely misfolded proteins that are not bound by α-crystallin but accumulate and have a direct toxic effect on lens cells, resulting in early onset cataracts.
Publicações recentes
Hereditary cataract in the Bengal cat in Poland.
Human βA3/A1-crystallin splicing mutation causes cataracts by activating the unfolded protein response and inducing apoptosis in differentiating lens fiber cells.
📚 EuropePMCmostrando 3
A novel ADAMTSL4 compound heterozygous mutation in isolated ectopia lentis: a case report and review of the literature.
Journal of medical case reportsHereditary cataract in the Bengal cat in Poland.
BMC veterinary researchHuman βA3/A1-crystallin splicing mutation causes cataracts by activating the unfolded protein response and inducing apoptosis in differentiating lens fiber cells.
Biochimica et biophysica actaAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Catarata das suturas de início precoce.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Catarata das suturas de início precoce
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A novel ADAMTSL4 compound heterozygous mutation in isolated ectopia lentis: a case report and review of the literature.
- Hereditary cataract in the Bengal cat in Poland.
- Human βA3/A1-crystallin splicing mutation causes cataracts by activating the unfolded protein response and inducing apoptosis in differentiating lens fiber cells.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:98985(Orphanet)
- MONDO:0020372(MONDO)
- GARD:16885(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q27674894(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
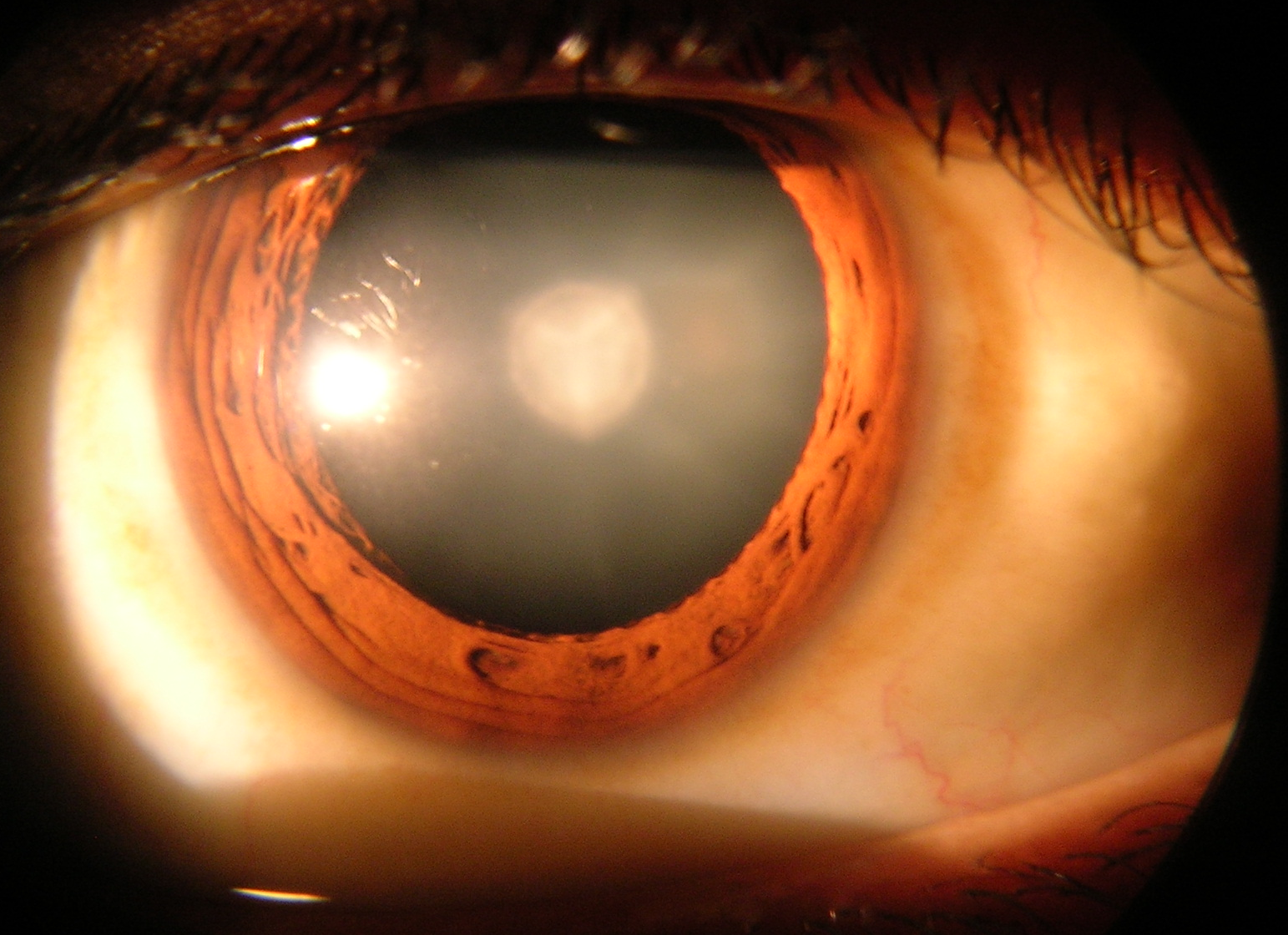
Catarata das suturas de início precoce
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata