Catarata é uma área opaca no cristalino do olho que prejudica a visão. As cataratas geralmente se desenvolvem lentamente e podem afetar um ou ambos os olhos. Os sintomas podem incluir cores desbotadas, visão embaçada ou dupla, halos ao redor de luzes, problemas com luzes fortes e dificuldade para enxergar à noite. Isso pode resultar em dificuldade para dirigir, ler e reconhecer rostos. A visão precária causada pela catarata também pode resultar em um risco aumentado de quedas e depressão. Em 2020, a catarata causou 39.
Introdução
O que você precisa saber de cara
Visão geral
A catarata subcapsular posterior de início precoce é uma condição rara que afeta a transparência do cristalino, a lente natural do olho. Nessa doença, a opacidade se forma na parte de trás do cristalino (região subcapsular posterior) e surge em idade mais jovem do que o esperado para cataratas relacionadas ao envelhecimento. A visão pode ficar embaçada ou com ofuscamento, especialmente em ambientes com luz forte.[1]
Sinais e sintomas
Os principais sintomas incluem diminuição progressiva da visão, dificuldade para enxergar em luz intensa (fotofobia) e sensação de ofuscamento. A catarata subcapsular posterior costuma afetar mais a visão de perto e a leitura, e pode piorar rapidamente. Como a opacidade fica na parte posterior do cristalino, os sintomas podem ser desproporcionais ao tamanho da lesão.[1]
Causas genéticas
A catarata subcapsular posterior de início precoce pode ser causada por variantes (mutações) em pelo menos cinco genes: EPHA2, CHMP4B, UNC45B, LEMD2 e CRYBB2. Esses genes fornecem instruções para proteínas importantes para a manutenção da transparência e da estrutura do cristalino. Alterações nesses genes podem levar ao acúmulo de proteínas anormais e à opacificação do cristalino.[1][3]
Diagnóstico
O diagnóstico é feito por um oftalmologista por meio do exame de lâmpada de fenda, que permite visualizar a opacidade na região subcapsular posterior do cristalino. Exames genéticos podem ajudar a confirmar a causa hereditária. Atualmente, existem 10 testes genéticos disponíveis para essa condição, e 111 variantes diferentes já foram registradas no banco de dados ClinVar.[1][3]
Tratamento e manejo
O tratamento padrão para catarata que prejudica a visão é a cirurgia de remoção do cristalino opaco e implante de lente intraocular. A decisão sobre o momento da cirurgia deve ser individualizada, considerando o impacto na qualidade de vida e a idade do paciente. Não existem medicamentos aprovados especificamente para tratar ou reverter a catarata subcapsular posterior de início precoce.[1]
Prognóstico e qualidade de vida
Com o tratamento cirúrgico adequado, a maioria das pessoas recupera uma visão funcional satisfatória. No entanto, a condição pode impactar atividades diárias como dirigir, ler e reconhecer rostos, especialmente se não for tratada. O acompanhamento oftalmológico regular é importante para monitorar a progressão e planejar a intervenção no momento certo.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Catarata é uma área opaca no cristalino do olho que prejudica a visão. As cataratas geralmente se desenvolvem lentamente e podem afetar um ou ambos os olhos. Os sintomas podem incluir cores desbotadas, visão embaçada ou dupla, halos ao redor de luzes, problemas com luzes fortes e dificuldade para enxergar à noite. Isso pode resultar em dificuldade para dirigir, ler e reconhecer rostos. A visão precária causada pela catarata também pode resultar em um risco aumentado de quedas e depressão. Em 2020, a catarata causou 39.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A catarata subcapsular posterior de início precoce é uma condição rara que afeta a transparência do cristalino, a lente natural do olho. Nessa doença, a opacidade se forma na parte de trás do cristalino (região subcapsular posterior) e surge em idade mais jovem do que o esperado para cataratas relacionadas ao envelhecimento. A visão pode ficar embaçada ou com ofuscamento, especialmente em ambientes com luz forte.[1]
Sinais e sintomas
Os principais sintomas incluem diminuição progressiva da visão, dificuldade para enxergar em luz intensa (fotofobia) e sensação de ofuscamento. A catarata subcapsular posterior costuma afetar mais a visão de perto e a leitura, e pode piorar rapidamente. Como a opacidade fica na parte posterior do cristalino, os sintomas podem ser desproporcionais ao tamanho da lesão.[1]
Causas genéticas
A catarata subcapsular posterior de início precoce pode ser causada por variantes (mutações) em pelo menos cinco genes: EPHA2, CHMP4B, UNC45B, LEMD2 e CRYBB2. Esses genes fornecem instruções para proteínas importantes para a manutenção da transparência e da estrutura do cristalino. Alterações nesses genes podem levar ao acúmulo de proteínas anormais e à opacificação do cristalino.[1][3]
Diagnóstico
O diagnóstico é feito por um oftalmologista por meio do exame de lâmpada de fenda, que permite visualizar a opacidade na região subcapsular posterior do cristalino. Exames genéticos podem ajudar a confirmar a causa hereditária. Atualmente, existem 10 testes genéticos disponíveis para essa condição, e 111 variantes diferentes já foram registradas no banco de dados ClinVar.[1][3]
Tratamento e manejo
O tratamento padrão para catarata que prejudica a visão é a cirurgia de remoção do cristalino opaco e implante de lente intraocular. A decisão sobre o momento da cirurgia deve ser individualizada, considerando o impacto na qualidade de vida e a idade do paciente. Não existem medicamentos aprovados especificamente para tratar ou reverter a catarata subcapsular posterior de início precoce.[1]
Prognóstico e qualidade de vida
Com o tratamento cirúrgico adequado, a maioria das pessoas recupera uma visão funcional satisfatória. No entanto, a condição pode impactar atividades diárias como dirigir, ler e reconhecer rostos, especialmente se não for tratada. O acompanhamento oftalmológico regular é importante para monitorar a progressão e planejar a intervenção no momento certo.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
5 genes identificados com associação a esta condição.
Receptor tyrosine kinase which binds promiscuously membrane-bound ephrin-A family ligands residing on adjacent cells, leading to contact-dependent bidirectional signaling into neighboring cells. The signaling pathway downstream of the receptor is referred to as forward signaling while the signaling pathway downstream of the ephrin ligand is referred to as reverse signaling. Activated by the ligand ephrin-A1/EFNA1 regulates migration, integrin-mediated adhesion, proliferation and differentiation
Cell membraneCell projection, ruffle membraneCell projection, lamellipodium membraneCell junction, focal adhesion
Cataract 6, multiple types
An opacification of the crystalline lens of the eye that frequently results in visual impairment or blindness. Opacities vary in morphology, are often confined to a portion of the lens, and may be static or progressive. CTRCT6 includes posterior polar and age-related cortical cataracts, among others. Posterior polar cataract is a subcapsular opacity, usually disk-shaped, located at the back of the lens. Age-related cortical cataract is a developmental punctate opacity restricted to the cortex. The cataract is white or cerulean, increases in number with age, but rarely affects vision.
Probable core component of the endosomal sorting required for transport complex III (ESCRT-III) which is involved in multivesicular bodies (MVBs) formation and sorting of endosomal cargo proteins into MVBs. MVBs contain intraluminal vesicles (ILVs) that are generated by invagination and scission from the limiting membrane of the endosome and mostly are delivered to lysosomes enabling degradation of membrane proteins, such as stimulated growth factor receptors, lysosomal enzymes and lipids. The M
Cytoplasm, cytosolLate endosome membraneMidbodyNucleus envelope
Cataract 31, multiple types
An opacification of the crystalline lens of the eye that frequently results in visual impairment or blindness. Opacities vary in morphology, are often confined to a portion of the lens, and may be static or progressive. In general, the more posteriorly located and dense an opacity, the greater the impact on visual function. CTRCT31 includes posterior polar, progressive posterior subcapsular, nuclear, and anterior subcapsular cataracts.
Acts as a co-chaperone for HSP90 and is required for proper folding of the myosin motor domain. Plays a role in sarcomere formation during muscle cell development. Is necessary for normal early lens development
Cytoplasm, myofibril, sarcomere, Z lineCytoplasm, myofibril, sarcomere, A bandCytoplasm, perinuclear regionCytoplasm, cytosol
Cataract 43
An opacification of the crystalline lens of the eye that frequently results in visual impairment or blindness. Opacities vary in morphology, are often confined to a portion of the lens, and may be static or progressive. In general, the more posteriorly located and dense an opacity, the greater the impact on visual function.
Nuclear lamina-associated inner nuclear membrane protein that is involved in nuclear structure organization, maintenance of nuclear envelope (NE) integrity and NE reformation after mitosis (PubMed:16339967, PubMed:17097643, PubMed:28242692, PubMed:32494070). Plays a role as transmembrane adapter for the endosomal sorting complexes required for transport (ESCRT), and is thereby involved in ESCRT-mediated NE reformation (PubMed:28242692, PubMed:32494070). Promotes ESCRT-mediated NE closure by recr
Nucleus inner membraneNucleus envelopeCytoplasm, cytoskeleton, spindle
Cataract 46, juvenile-onset, with or without arrhythmic cardiomyopathy
A form of cataract, an opacification of the crystalline lens of the eye that frequently results in visual impairment or blindness. Opacities vary in morphology, are often confined to a portion of the lens, and may be static or progressive. In general, the more posteriorly located and dense an opacity, the greater the impact on visual function. CTRCT46 can be associated with variable onset of a severe form of arrhythmic cardiomyopathy resulting in sudden cardiac death.
Crystallins are the dominant structural components of the vertebrate eye lens
Cataract 3, multiple types
An opacification of the crystalline lens of the eye that frequently results in visual impairment or blindness. Opacities vary in morphology, are often confined to a portion of the lens, and may be static or progressive. CTRCT3 includes congenital cerulean and sutural cataract with punctate and cerulean opacities, among others. Cerulean cataract is characterized by peripheral bluish and white opacifications organized in concentric layers with occasional central lesions arranged radially. The opacities are observed in the superficial layers of the fetal nucleus as well as the adult nucleus of the lens. Involvement is usually bilateral. Visual acuity is only mildly reduced in childhood. In adulthood, the opacifications may progress, making lens extraction necessary. Histologically the lesions are described as fusiform cavities between lens fibers which contain a deeply staining granular material. Although the lesions may take on various colors, a dull blue is the most common appearance and is responsible for the designation cerulean cataract. Sutural cataract with punctate and cerulean opacities is characterized by white opacification around the anterior and posterior Y sutures, and grayish and bluish, spindle shaped, oval punctate and cerulean opacities of various sizes arranged in lamellar form. The spots are more concentrated towards the peripheral layers and do not delineate the embryonal or fetal nucleus. Phenotypic variation with respect to the size and density of the sutural opacities as well as the number and position of punctate and cerulean spots is observed among affected subjects.
Variantes genéticas (ClinVar)
111 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
24 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Catarata subcapsular posterior de início precoce
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Senescence-Driven IL-17A Inflammatory Circuit Promotes Epithelial-Mesenchymal Transition (EMT) and Progression in Age-Related Posterior Subcapsular Cataracts.
Cataract is a leading cause of visual impairment worldwide, and its prevalence is increasing with population aging. The majority are age-related cataracts (ARC), clinically classified into nuclear, cortical, and posterior subcapsular cataracts (PSC) by opacity location. Mechanisms of cataractogenesis remain incompletely understood. While cortical and nuclear cataracts are largely attributed to crystallin aggregation, such protein-centric mechanisms fail to explain the early onset and axial location of PSC. Morphologically, PSC resembles posterior capsule opacification (PCO), a secondary cataract driven by epithelial-mesenchymal transition (EMT) of lens epithelial cells (LECs), suggesting LEC-EMT may also contribute to PSC. Using clinically stratified human lens samples, we confirmed EMT marker expression across PSC subtypes. Transcriptomic profiling revealed that LECs, the lens's sole metabolically active cells, in age-related PSC (ARC-PSC), compared with age-related nuclear cataract (ANC) and clear lenses, exhibit elevated EMT signatures tightly linked to senescence-associated inflammatory signaling. Aqueous humor (AqH) profiling demonstrated a pro-inflammatory milieu across PSC subtypes, with IL-17A uniquely elevated in ARC-PSC, consistent with transcriptomic findings. Integrated analyses support a model in which senescence functions as an upstream driver, whereby senescent LECs release SASP factors, including IL-17A, that activate NF-κB signaling to amplify inflammation, reinforce senescence, and drive EMT. In vitro, senolysis and IL-17A blockade disrupted this loop, attenuating senescence- and EMT-associated phenotypes. Collectively, our study demonstrates that senescent LECs sustain an IL-17A-NF-κB circuit that drives EMT and accelerates ARC-PSC progression, positioning ARC-PSC as a unique human context to study senescence-induced epithelial remodeling in aging tissues.
A new genotype of the IDH3A gene causes retinitis pigmentosa, generating functional dyschromatopsia from early childhood.
We report the case of a 42-year-old Venezuelan woman with childhood-onset autosomal recessive retinitis pigmentosa type 90 (RP90), presenting an unusual and distinctive clinical phenotype characterized by macular pseudocoloboma, very early-onset acquired color vision disorder progressing to severe functional dyschromatopsia, and early-onset severe posterior subcapsular cataracts. Her affected brother exhibited a similar phenotype, while her parents and the other two siblings remained unaffected. Massive parallel sequencing identified two novel IDH3A variants: c.127G>T (chr15:78449926 G>T; no dbSNP entry) and c.419T>C (chr15:78454052T>C; no dbSNP entry), in compound heterozygosity and confirmed to be in trans location. Both missense variants, absent from population databases, were predicted to be deleterious by multiple in silico tools and are located in critical domains involved in enzymatic complex stability, catalytic activity and subunit interactions. This case reinforces the association between IDH3A mutations and RP90, corroborates key phenotypic features described in limited published reports-the presence of macular pseudocoloboma-and expands the mutational spectrum of the gene. Moreover, it highlights the role of mitochondrial metabolism in photoreceptor degeneration and proposes a link between them. Our findings underscore the need for functional studies to elucidate the pathogenic mechanisms underlying IDH3A-related retinopathies.
Ocular Findings as the Most Striking Manifestation of a SMAD3 Variant.
Loeys-Dietz syndrome (LDS) is a heritable connective tissue disorder with variable expressivity. It is a multisystemic condition mainly characterized by a propensity for arterial aneurysms and dissections, skeletal manifestations, hypertelorism, bifid uvula, craniosynostosis, and cutaneous features. While LDS has significant clinical overlap with other heritable connective tissue disorders such as Marfan syndrome, the ocular manifestations of LDS have not yet been well characterized. We describe an individual with a variant in SMAD3 with a predominantly ocular phenotype and only mild arterial manifestations. The proband presented with early-onset high myopia (-25 diopters, bilaterally) and bilateral posterior subcapsular cataracts. Ocular phenotype also included esotropia, retinal hemorrhage, glaucoma, bilateral posterior staphylomas, and keratoconjunctivitis sicca. Extensive vascular imaging identified a right internal carotid artery dissection and mild tortuosity of numerous arteries. A molecular gene panel for heritable connective tissue disorders identified a heterozygous SMAD3 c.221G > T; p.(Arg74Leu) variant, initially reported as a variant of uncertain significance and subsequently re-interpreted as likely pathogenic. Other missense variants at this residue have been reported as pathogenic for LDS. High-grade myopia, premature cataracts, and premature glaucoma are part of the phenotypic spectrum in LDS caused by SMAD3 variants. Inclusion of SMAD3 in molecular gene panels for early-onset high-grade myopia should be considered.
A Rare Vitreoretinal Degenerative Disorder: Goldmann-Favre Syndrome Complicated with Choroidal Neovascularization in a Pediatric Patient.
Goldmann-Favre syndrome (GFS) is a rare vitreoretinal degenerative disorder caused by mutations in the NR2E3 gene located on the short arm of chromosome 15. This condition, inherited in an autosomal recessive manner, was first described by Favre in two siblings, with Ricci later confirming its hereditary pattern. In GFS, rod photoreceptors are essentially replaced by S-cone photoreceptors. Enhanced S-Cone Syndrome (ESCS) and Goldmann-Favre syndrome are two distinct entities within the spectrum of retinal degenerative diseases, both caused by mutations in the NR2E3 gene. Despite sharing a common genetic basis, these conditions exhibit significantly different clinical phenotypes. ESCS is characterized by an excessive number of S-cones (blue-sensitive cones) with degeneration of rods and L-/M-cones, leading to increased sensitivity to blue light and early-onset night blindness. In contrast, GFS is considered a more severe form of ESCS, involving additional features such as retinal schisis, vitreous degeneration, and more pronounced visual impairment. GFS typically manifests in the first decade of life as night blindness (nyctalopia) and progressive visual acuity impairment. The clinical features include degenerative vitreous changes such as liquefaction, strands, and bands, along with macular and peripheral retinoschisis, posterior subcapsular cataract, atypical pigmentary dystrophy, and markedly abnormal or nondetectable electroretinograms (ERGs). Although peripheral retinoschisis is more common in GFS, central retinoschisis may also occur. Despite the consistent genetic basis, the phenotype of GFS can vary significantly among individuals. The differential diagnosis should consider diseases within the retinal degenerative spectrum, including retinitis pigmentosa, congenital retinoschisis, and secondary pigmentary retinopathy.
Case Series on Autosomal Recessive Non-Syndromic Retinitis Pigmentosa Caused by POMGNT1 Mutations with a Report of a New Variant.
Recessive Protein O-linked-mannose beta-1,2-N-acetylglucosaminyltransferase 1 (POMGNT1) mutations can cause early onset muscle-eye-brain disease but have also more recently been associated with non-syndromic Retinitis Pigmentosa. In this case series, we describe three sisters affected by non-syndromic autosomal recessive POMGNT1 retinopathy with a report of a new variant. The three patients received care at West Virginia University Eye Institute, including full ophthalmic examination with additional fundus imaging, optical coherence tomography (OCT), electroretinogram (ERG), and visual field testing. Diagnostic panel testing of 330 genes was also obtained. The proband was seen for cataract evaluation at age 42, and her fundus examination was suggestive of retinitis pigmentosa. Her oldest sister had been treated for acute anterior uveitis with retinal vasculitis. Another sister was diagnosed with multiple sclerosis (MS) and peripheral retinal degeneration. Posterior subcapsular cataracts were diagnosed between age 42 and 55 in all three sisters, each with constricted fields with preserved central vision. We identified one pathogenic POMGNT1 variant (c.751 + 1G > A) and one likely pathogenic variant (c.1010T > C p.Ile337Thr) in all three sisters. A thorough family history and examination of the siblings with genotyping might have led to an earlier diagnosis of retinal inherited disease and avoidance of immunomodulatory treatment in the oldest sibling.
Publicações recentes
Ocular Findings as the Most Striking Manifestation of a SMAD3 Variant.
Association between single nucleotide polymorphisms in exon 3 of the alpha-A-crystallin gene and susceptibility to age-related cataract.
Efficacy and safety of ropivacaine HCl in peribulbar anaesthesia for cataract surgery in patients with myotonic dystrophy type 1.
[Neurofibromatosis type 2 in childhood: a clinical characterization].
Early onset steroid induced posterior subcapsular cataract in a patient with common variable immunodeficiency: case reports and review of literature.
📚 EuropePMCmostrando 11
Senescence-Driven IL-17A Inflammatory Circuit Promotes Epithelial-Mesenchymal Transition (EMT) and Progression in Age-Related Posterior Subcapsular Cataracts.
Aging cellA new genotype of the IDH3A gene causes retinitis pigmentosa, generating functional dyschromatopsia from early childhood.
Ophthalmic geneticsOcular Findings as the Most Striking Manifestation of a SMAD3 Variant.
American journal of medical genetics. Part AA Rare Vitreoretinal Degenerative Disorder: Goldmann-Favre Syndrome Complicated with Choroidal Neovascularization in a Pediatric Patient.
Diagnostics (Basel, Switzerland)Case Series on Autosomal Recessive Non-Syndromic Retinitis Pigmentosa Caused by POMGNT1 Mutations with a Report of a New Variant.
Journal of clinical medicineLEPREL1 -RELATED GIANT RETINAL TEAR DETACHMENTS MIMIC THE PHENOTYPE OF OCULAR STICKLER SYNDROME.
Retina (Philadelphia, Pa.)Association between single nucleotide polymorphisms in exon 3 of the alpha-A-crystallin gene and susceptibility to age-related cataract.
Ophthalmic geneticsEfficacy and safety of ropivacaine HCl in peribulbar anaesthesia for cataract surgery in patients with myotonic dystrophy type 1.
Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of MyologyPresenile cataract and its risk factors: A case control study.
Journal of family medicine and primary care[Neurofibromatosis type 2 in childhood: a clinical characterization].
Revista de neurologiaEarly onset steroid induced posterior subcapsular cataract in a patient with common variable immunodeficiency: case reports and review of literature.
European annals of allergy and clinical immunologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Catarata subcapsular posterior de início precoce.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Catarata subcapsular posterior de início precoce
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Senescence-Driven IL-17A Inflammatory Circuit Promotes Epithelial-Mesenchymal Transition (EMT) and Progression in Age-Related Posterior Subcapsular Cataracts.
- A new genotype of the IDH3A gene causes retinitis pigmentosa, generating functional dyschromatopsia from early childhood.
- Ocular Findings as the Most Striking Manifestation of a SMAD3 Variant.
- A Rare Vitreoretinal Degenerative Disorder: Goldmann-Favre Syndrome Complicated with Choroidal Neovascularization in a Pediatric Patient.
- Case Series on Autosomal Recessive Non-Syndromic Retinitis Pigmentosa Caused by POMGNT1 Mutations with a Report of a New Variant.
- Association between single nucleotide polymorphisms in exon 3 of the alpha-A-crystallin gene and susceptibility to age-related cataract.
- Efficacy and safety of ropivacaine HCl in peribulbar anaesthesia for cataract surgery in patients with myotonic dystrophy type 1.
- [Neurofibromatosis type 2 in childhood: a clinical characterization].
- Early onset steroid induced posterior subcapsular cataract in a patient with common variable immunodeficiency: case reports and review of literature.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:441447(Orphanet)
- MONDO:0018610(MONDO)
- GARD:21846(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55788216(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
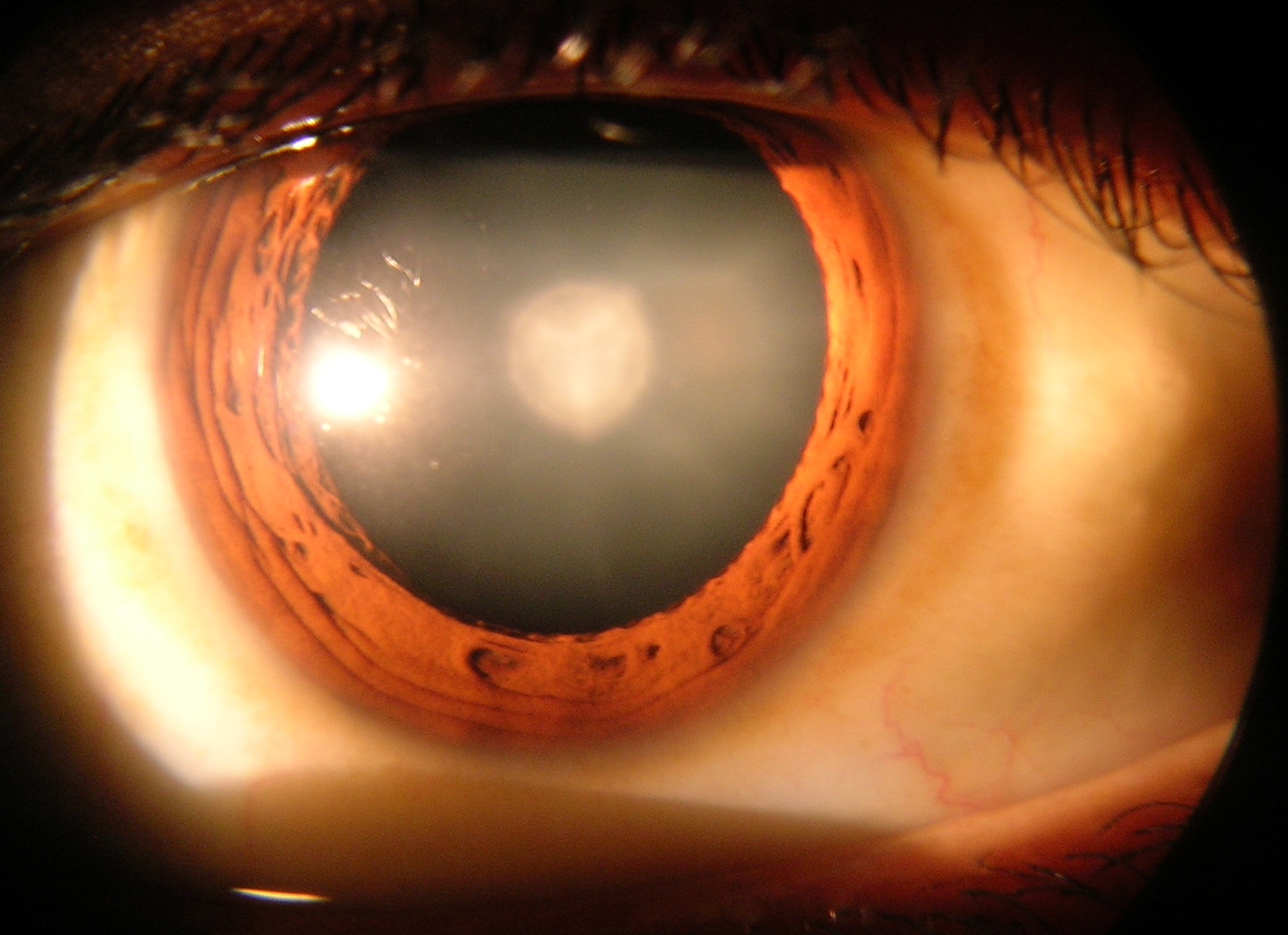
Catarata subcapsular posterior de início precoce
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata