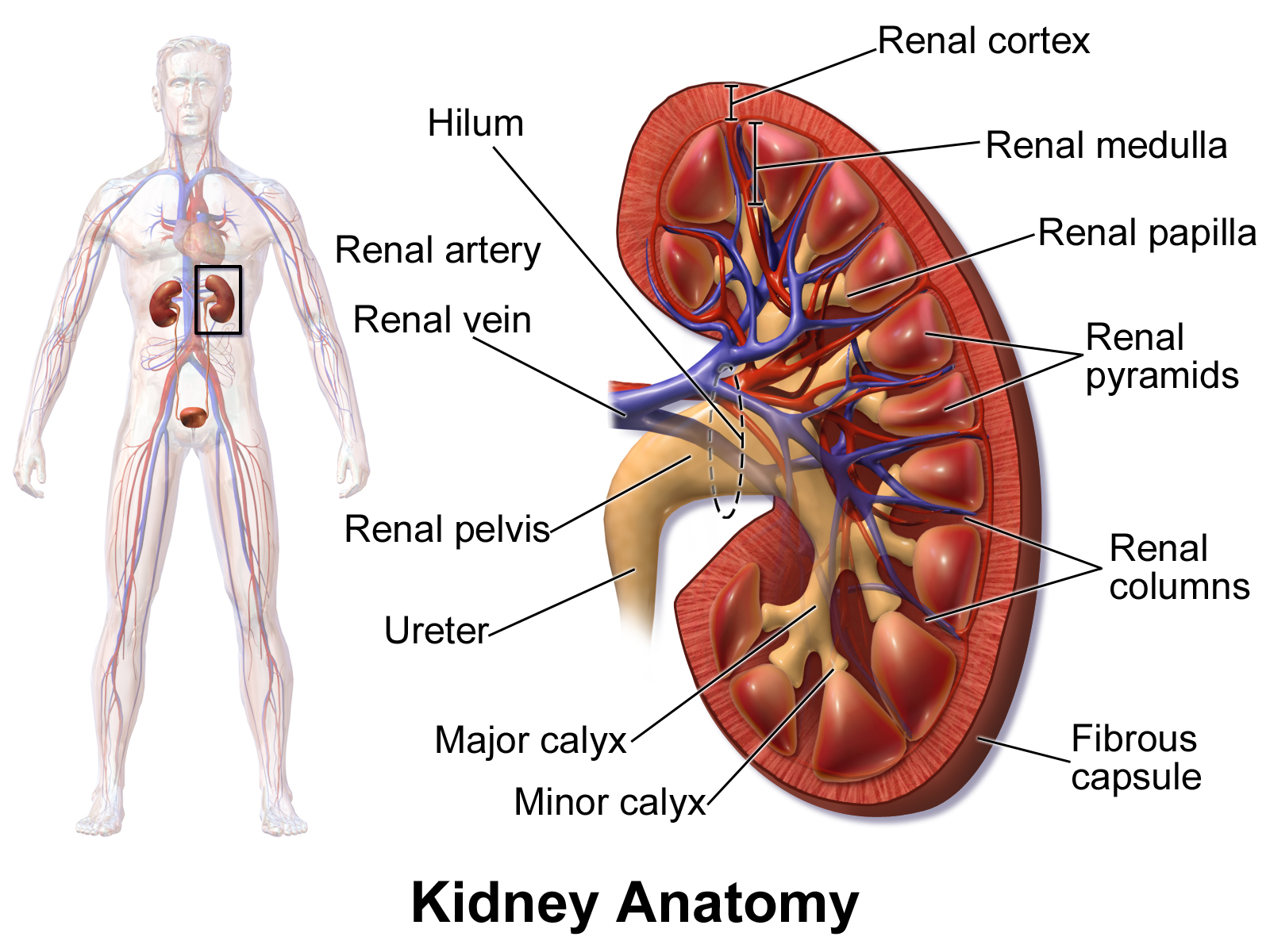
Displasia renal unilateral é um tipo de displasia renal (DR). É um defeito no desenvolvimento do sistema urinário em que um dos rins não se forma completamente ou se desenvolve de maneira anormal. A DR unilateral pode afetar apenas uma parte do rim e ter diferentes níveis de gravidade. A forma mais grave é quando o rim não se forma de jeito nenhum, condição chamada de aplasia renal.
Introdução
O que você precisa saber de cara
Displasia renal unilateral é um tipo de displasia renal (DR). É um defeito no desenvolvimento do sistema urinário em que um dos rins não se forma completamente ou se desenvolve de maneira anormal. A DR unilateral pode afetar apenas uma parte do rim e ter diferentes níveis de gravidade. A forma mais grave é quando o rim não se forma de jeito nenhum, condição chamada de aplasia renal.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant, Not applicable.
Transcription factor that binds to the inverted palindrome 5'-GTTAATNATTAAC-3' (PubMed:17924661, PubMed:7900999). Binds to the FPC element in the cAMP regulatory unit of the PLAU gene (By similarity). Transcriptional activity is increased by coactivator PCBD1 (PubMed:24204001)
Nucleus
Renal cysts and diabetes syndrome
An autosomal dominant disorder comprising non-diabetic renal disease resulting from abnormal renal development, and diabetes, which in some cases occurs earlier than age 25 years and is thus consistent with a diagnosis of maturity-onset diabetes of the young (MODY5). The renal disease is highly variable and includes renal cysts, glomerular tufts, aberrant nephrogenesis, primitive tubules, irregular collecting systems, oligomeganephronia, enlarged renal pelves, abnormal calyces, small kidney, single kidney, horseshoe kidney, and hyperuricemic nephropathy. Affected individuals may also have abnormalities of the genital tract.
Variantes genéticas (ClinVar)
534 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
6 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Displasia renal, unilateral
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
2 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Unilateral Multicystic Dysplastic Kidney in a Fetus Associated With Parental Genetic and Environmental Risk Factors: A Case Report.
Multicystic dysplastic kidney (MCDK) is a congenital renal anomaly identified on prenatal ultrasound. They often arise sporadically and unilaterally. Our case involves an isolated unilateral MCDK in a fetus born to a mother with generalized anxiety disorder (GAD), obsessive-compulsive disorder (OCD), and gastroesophageal reflux disease (GERD), with the father having chronic occupational lead exposure and a congenital disorder of glycosylation type 1A (PMM2-CDG). Our case highlights the multifactorial etiology of renal dysplasia and its potential role of glycosylation defects and environmental toxicity in abnormal kidney development. Contributions from genetic, environmental, and metabolic influences during nephrogenesis contribute to MCDK.
Prevalence, clinical profile, and associated anomalies with women with Mayer-Rokitansky-Küster-Hauser syndrome in a tertiary care center: A cross-sectional study.
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome is a rare congenital anomaly of the Müllerian ducts and represents the second most common cause of primary amenorrhea, accounting for 10%-15% of cases. Despite its significance, limited data exist regarding its clinical profile and associated anomalies in the Indian population. This study aims to determine the prevalence of MRKH syndrome among women presenting with primary amenorrhea at a tertiary care center in South India and to describe their clinical profiles and associated anomalies using the Vagina Cervix Uterus Adnexa-associated Malformation (VCUAM) classification system. A retrospective study was conducted over 15 years (January 2008 to December 2022) including women diagnosed with MRKH syndrome based on inclusion criteria: primary amenorrhea, normal secondary sexual characteristics, 46-XX karyotype, and normal serum follicle-stimulating hormone levels. Data were extracted from medical records and analyzed using SPSS v25.0. Out of 340 women with primary amenorrhea, 181 (60%) were diagnosed with MRKH syndrome. The mean age at presentation was 21 years. The predominant complaint was non-attainment of menarche (66.8%), with 16.5% reporting cyclical abdominal pain. Type 1 MRKH was most common (78.9%), followed by Müllerian duct aplasia-renal agenesis-cervicothoracic somite dysplasia (MURCS) association (16.5%). Renal anomalies (15.5%) were the most frequent extragenital malformations. All women had vaginal and cervical agenesis (V5bC2b). Uterine anomalies included bilateral aplasia (89.1%), unilateral aplasia (0.6%), and hypoplasia (10.5%). MRKH syndrome is a significant cause of primary amenorrhea, with notable extragenital anomalies, especially renal. Systematic evaluation using the VCUAM classification enables comprehensive assessment, aiding in individualized and multidisciplinary care strategies.
Prenatal diagnosis of fetuses with renal abnormalities: a retrospective analysis of 329 Chinese cases.
There is no clear guidance on prenatal diagnostic testing strategies for congenital renal anomalies. Therefore, this study aims to investigate the retrospective analysis of ultrasound and genetic diagnostic results in cases of fetal renal abnormalities and to establish genotype-phenotype correlations. A total of 329 fetuses with renal abnormalities that underwent prenatal diagnostic testing from January 2020 to April 2023 were recruited in this study. These cases were classified into 11 subgroups based on their ultrasound diagnosis. All cases underwent chromosomal microarray analysis (CMA) or copy number variation sequencing (CNV-seq), with subsequent whole exome sequencing (WES) conducted on select CMA/CNV-seq negative cases, subject to parental consent for further testing targeting monogenic variations. Of the 329 cases analyzed, CMA/CNV-seq detected chromosomal abnormalities in 31 cases, with a detection rate of 9.4% (31/329). The most common abnormality was 17q12 deletion, accounting for 29% of the positive cases (9/31) and 2.7% of the total cases (9/329). WES was conducted on 76 cases (76/298, 25.5%), revealing 16 monogenic variants, and 2 CNVs in 12 cases (15.8%). An overall positive diagnostic yield of 13.1% (43/329) was obtained in the pipeline of combinational CMA/CNV-seq and WES analysis. Ciliary genes (TMEM67, NPHP3, CEP290, BBS2, and TTC8) were frequently implicated by WES. Several genotype-phenotype correlations emerged, including (1) hyperechogenic kidneys associated with 17q12 deletion, (2) renal dysplasia, renal cysts, hydronephrosis, ectopic kidney, and renal duplication with chromosomal abnormalities, (3) unilateral renal agenesis and polycystic kidneys with monogenic variants. This study reveals genotype-phenotype correlations in fetal renal abnormalities, informing prenatal counseling regarding diagnostic testing options and expected outcomes.
Case Report: Diagnostic overlap of OHVIRA syndrome and Gartner duct cyst: challenges in imaging and management.
This case series explores the diagnostic overlap between OHVIRA syndrome (obstructed hemivagina and ipsilateral renal anomaly) and Gartner duct cyst (GDC) with ipsilateral renal dysplasia in two prepubertal girls. Both cases exhibited similar imaging features on CT and MRI-including unilateral renal agenesis, ectopic ureter with abnormal insertion, cystic lesions posterolateral to the bladder- which led to significant diagnostic challenges. Despite differing in pathology, the overlap in embryologic origins and imaging findings made differentiation difficult. This series underscores the importance of comprehensive imaging and a multidisciplinary approach for accurate diagnosis. Current management strategies are discussed, highlighting the need for individualized treatment plans in prepubertal patients with complex genitourinary anomalies. Cat eye syndrome (CES), also known as Schmid-Fraccaro syndrome, is a rare genetic disorder named for the vertical iris coloboma observed in some affected individuals. The condition is classically characterized by a triad of features—iris coloboma, anal atresia, and preauricular pits or tags. However, CES can also involve a range of abnormalities affecting the neurodevelopmental, ocular, auricular, nasal, cardiovascular, gastrointestinal, and urogenital systems (see Image. Schematic Diagram Showing Iris Coloboma in Cat Eye Syndrome). The clinical presentation of CES is variable, with differences in affected organ systems, prognosis, genetics, and heritability among individuals. CES is a rare chromosomal disorder first described in the early 1960s by Schmid and Fraccaro. This condition is characterized by a partial tetrasomy or trisomy of chromosome 22q11.1-q11.2. The name "cat eye" originates from ocular colobomas—iris defects—present in about half of affected individuals, which give the pupil a distinctive keyhole or cat-eye appearance. Although ocular coloboma is the eponymous hallmark, CES is fundamentally a multisystem genomic disorder with highly variable expressivity, spanning a spectrum from nearly asymptomatic to severe anomalies across ocular, cardiac, renal, gastrointestinal, skeletal, and neurodevelopmental domains. The association between ocular coloboma and anal atresia was first described by Haab in 1878. The genetic alteration is due to a small supernumerary marker chromosome (sSMC), which was first described in 1965. Schachenmann et al reported 3 pediatric patients and 1 patient’s mother who carried an additional, abnormally small chromosome featuring a submedian centromere, while the rest of the karyotype appeared normal. This sSMC contains the CES critical region (CESCR), located within the proximal portion of chromosome 22q11.2, between the centromere and the LCR22-A region. Additional genetic conditions related to chromosome 22 include the oculo-auriculo-vertebral spectrum (OAVS), DiGeorge syndrome, and mosaic trisomy 22. At the genetic level, CES arises from a supernumerary marker chromosome, often dicentric, composed of material from chromosome 22. In approximately 90% of cases, this marker contains 2 extra copies of the proximal 22q11 region (tetrasomy), while a smaller proportion exhibits an additional copy (trisomy). The critical region encompasses approximately 1.5 to 2 Mb and includes multiple dosage-sensitive genes whose overexpression is believed to contribute to the diverse phenotypic features of CES. Molecular cytogenetic techniques, such as fluorescence in situ hybridization (FISH), array comparative genomic hybridization (aCGH), and, more recently, genome-wide single-nucleotide polymorphism (SNP) microarrays, have replaced traditional karyotyping for precise delineation of the supernumerary chromosome and identification of the breakpoints. This high-resolution genomic mapping is crucial for definitive diagnosis, genotype-phenotype correlations, and recurrence-risk counseling. Clinically, CES is remarkably heterogeneous. The classic triad comprises iris coloboma, preauricular skin tags or pits, and anal atresia or other anorectal malformations. However, no single feature is universally present. Iris coloboma appears in 40% to 60% of cases, preauricular anomalies in up to 70%, and anorectal malformations in about 30% to 50%. Cardiac defects, most commonly total or partial atrioventricular septal defects and tetralogy of Fallot, occur in approximately half of patients and are a major contributor to early morbidity and mortality. Renal anomalies, reported in 20% to 40% of cases, may include unilateral renal agenesis, duplex collecting systems, hydronephrosis, and vesicoureteral reflux. Skeletal abnormalities range from vertebral segmentation defects to limb anomalies. Otolaryngological manifestations may include hearing loss from middle-ear dysplasia. Less commonly, gastrointestinal anomalies beyond anorectal malformations—such as duodenal atresia or Hirschsprung disease—have also been documented. Neurodevelopmental outcomes in CES vary widely. Although some children achieve developmental milestones within normal limits, others present with global developmental delay, intellectual disability, or features consistent with autism spectrum disorder. Hypotonia during infancy and feeding difficulties—often related to underlying gastrointestinal anomalies—may further compromise early growth. Growth parameters can be affected, with some patients exhibiting short stature or failure to thrive; however, many ultimately achieve normal height and weight. Behavioral phenotypes—such as attention-deficit/hyperactivity disorder (ADHD) and anxiety disorders—have also been reported, emphasizing the importance of comprehensive developmental and psychological assessment. Ophthalmic manifestations in CES extend beyond the iris coloboma. Additional anomalies such as chorioretinal colobomas, microphthalmia, cataracts, microcornea, and strabismus can contribute to visual impairment. A comprehensive ophthalmologic evaluation includes slit-lamp biomicroscopy to assess anterior segment abnormalities, indirect ophthalmoscopy for posterior segment examination, and optical coherence tomography (OCT), when available, to delineate the extent of colobomatous defects. Early detection and management of refractive errors, amblyopia, and strabismus are essential to support optimal visual development. Surgical intervention for coloboma is rarely indicated and is typically reserved for cases involving severe aniridia-like photophobia or significant cosmetic concerns (see Image. Schematic Diagram Showing Chorioretinal Coloboma in Cat Eye Syndrome). The management of CES depends on the organ systems involved and the severity of associated malformations. Given the significant clinical heterogeneity, an individualized, interprofessional approach is essential. This activity outlines the genetic and phenotypic spectrum of CES and outlines strategies for tailoring medical care to each patient’s needs. Cardiac evaluation at diagnosis is mandatory. Echocardiography within the first weeks of life is essential for detecting structural heart disease; in moderate-to-severe cases, surgical repair during infancy may be lifesaving. Long-term cardiology follow-up is critical to monitor for residual defects, arrhythmias, and pulmonary hypertension. Similarly, early renal ultrasonography is recommended to identify anatomical anomalies, guide urologic management, and prevent complications such as hypertension or renal insufficiency. Gastroenterological and colorectal management primarily focuses on anorectal malformations. Posterior sagittal anorectoplasty (PSARP) is the standard repair for imperforate anus, with timing and technical details tailored to the patient’s specific anatomy and overall health. Nutritional support—ranging from gavage or gastrostomy feeding in neonates to dietary modifications in older children—is essential, especially when gastrointestinal motility disorders or malabsorption are present. Audiologic and otologic care begins with newborn hearing screening. Conductive hearing loss due to middle ear anomalies may require interventions such as tympanostomy tubes or myringotomy. Speech therapy and educational support, tailored to the child’s developmental needs, are essential for optimizing communication outcomes. Genetic counseling for families includes discussion of recurrence risk, which is generally low (<1%) in de novo cases but higher in familial instances when a parent carries the small supernumerary marker chromosome in a balanced form. Secondary complications may include endocrine disorders, particularly growth hormone deficiency and thyroid dysfunction, necessitating regular endocrinologic screening. Orthopedic evaluations focus on detecting scoliosis and limb-length discrepancies. Dental and orthodontic assessments help identify malocclusion and enamel hypoplasia. Psychosocial support for families—including referrals to patient advocacy groups and peer support networks—promotes coping strategies and shared experiences. From a research perspective, CES provides valuable insights into gene dosage effects in contiguous-gene syndromes. The 22q11 region implicated in CES overlaps with that of DiGeorge syndrome (22q11.2 deletion), yet their phenotypes differ, reflecting divergent consequences of haploinsufficiency versus gene overexpression. Current studies focus on elucidating the roles of candidate genes such as CECR1 (which encodes adenosine deaminase 2) and CECR2 (involved in chromatin remodeling) in contributing to CES manifestations. Animal models with targeted duplications of the 22q11 region are under development to investigate relevant developmental pathways. Additionally, next-generation sequencing techniques show promise in detecting cryptic rearrangements and refining genotype–phenotype correlations, ultimately improving prognostic accuracy and identifying potential therapeutic targets. In summary, CES is a complex, multisystem chromosomal disorder. While its hallmark features include ocular coloboma, ear anomalies, and anorectal malformations, the condition also encompasses a wider phenotypic spectrum affecting the cardiac, renal, skeletal, neurodevelopmental, and endocrine systems. Accurate diagnosis relies on high-resolution cytogenetic and molecular techniques. Effective management requires coordinated multidisciplinary care, involving specialties from neonatology and cardiology to ophthalmology, urology, and developmental pediatrics. As advances in molecular genetics continue, they will enhance personalized prognostic counseling and enable the development of targeted therapies, ultimately improving outcomes for individuals and families affected by this rare but informative genomic syndrome.
Unilateral Inguinal Swelling in a Young Female: An Unusual Presentation of MURCS.
Inguinal hernia in females is an uncommon entity. While most patients present in infancy or early in childhood, only a few cases are diagnosed in adulthood. Most cases of inguinal hernia have small bowel or omentum as its content. Herniation of the ovary or fallopian tube is rare. In our case, an 18-year-old female presented to the outpatient department with unilateral inguinal swelling, which on imaging was found to be ovarian inguinal herniation. This prompted further evaluation. There was an associated absence of the uterus and left kidney, and congenital block vertebrae involving the cervical spine. On probing it was found that she had primary amenorrhoea with normal secondary sexual characteristics. All the findings led to the diagnosis of Mayer Rokitansky Kuster Hauser type II or Mullerian duct aplasia renal agenesis cervicothoracic somite dysplasia (MURCS) with unilateral inguinal ovarian herniation. Mullerian duct aplasia renal agenesis cervicothoracic somite dysplasia (MURCS) present a challenge as they require a multidisciplinary team including gynaecologist, surgeon and psychologist to preserve the ovarian function and help the patient counsel regarding the reproductive outcome and wade through the associated emotional stress. FREM1 autosomal recessive disorders include Manitoba oculotrichoanal (MOTA) syndrome, bifid nose with or without anorectal and renal anomalies (BNAR) syndrome, and isolated congenital anomalies of kidney and urinary tract (CAKUT). MOTA syndrome is characterized by an aberrant hairline (unilateral or bilateral wedge-shaped extension of the anterior hairline from the temple region to the ipsilateral eye) and anomalies of the eyes (widely spaced eyes, anophthalmia/microphthalmia and/or cryptophthalmos, colobomas of the upper eyelid, and corneopalpebral synechiae), nose (bifid or broad nasal tip), abdominal wall (omphalocele or umbilical hernia), and anus (stenosis and/or anterior displacement of the anal opening). The manifestations and degree of severity vary even among affected members of the same family. Growth and psychomotor development are normal. BNAR syndrome is characterized by a bifid or wide nasal tip, anorectal anomalies, and kidney malformations (e.g., renal agenesis, renal dysplasia). Typically, the eye manifestations of MOTA syndrome are absent. FREM1-related CAKUT has been reported in two boys from Macedonia with isolated CAKUT who had the same homozygous FREM1 pathogenic variants. The diagnosis of a FREM1 autosomal recessive disorder is established in a proband with biallelic pathogenic variants in FREM1 identified by molecular genetic testing. Treatment of manifestations: Intensive ocular lubrication to avoid exposure keratopathy before surgery is performed; release of synechiae between the eyelid and cornea; surgical intervention and/or prostheses for anophthalmia/microphthalmia and cryptophthalmos if warranted; supportive care for those with visual impairment. Rhinoplasty for notched ala nasi or bifid nose. Dilation for anal stenosis. Surgical closure of omphalocele; surgical or conservative management of umbilical hernia. Supportive treatment to preserve kidney function and electrolyte balance; dialysis and transplant if indicated in individuals with kidney failure. Psychosocial support and care coordination as needed. Surveillance: Assess kidney function in those with kidney disease with frequency per nephrologist; social work and family support at each visit. Phenotypes caused by biallelic FREM1 pathogenic variants – including MOTA syndrome, BNAR syndrome, and FREM1-related CAKUT – are inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a FREM1 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being a carrier, and a 25% chance of being unaffected and not a carrier. Once the FREM1 pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives and prenatal/preimplantation genetic testing are possible.
Publicações recentes
Transient neonatal diabetes mellitus as an early diagnostic clue to HNF1B-related disease - two case reports and a literature review.
Mayer-Rokitansky-Küster-Hauser Syndrome Associated With Diabetes Mellitus and Renal Anomalies in an Adolescent Girl: A Rare Case Report.
Adult Diagnosis of Solitary Kidney and Renal Dysplasia in a Male Born Prematurely as a Twin: A Case Report.
Meckel-Gruber syndrome: a rare and fatal congenital disorder (case report).
Aorto-Renal Dysplasia in Childhood: The Overlap of Neurofibromatosis Type 1 and Pediatric Renovascular Hypertension.
📚 EuropePMC1 artigos no totalmostrando 101
Unilateral Multicystic Dysplastic Kidney in a Fetus Associated With Parental Genetic and Environmental Risk Factors: A Case Report.
CureusPrevalence, clinical profile, and associated anomalies with women with Mayer-Rokitansky-Küster-Hauser syndrome in a tertiary care center: A cross-sectional study.
International journal of gynaecology and obstetrics: the official organ of the International Federation of Gynaecology and ObstetricsGenetic Analysis of Prenatal Renal Abnormalities in 17q12 Microdeletion Syndrome.
Maternal-fetal medicine (Wolters Kluwer Health, Inc.)Unmasking Secondary Hypertension: Renal Artery Stenosis Concealing the Diagnosis of Primary Hyperaldosteronism.
CureusPaediatric multicystic dysplastic kidney disease in Cape Town, South Africa.
BMC nephrologyMayer-Rokitansky-Küster-Hauser Syndrome: Where Does Gynaecological Pathology End and Renal Disease Begin? The Value of a Comprehensive View. Two Case Reports with Adult Onset Kidney Disease and A Review of the Literature.
Giornale italiano di nefrologia : organo ufficiale della Societa italiana di nefrologiaUnilateral Multicystic Dysplastic Kidney in an Infant: A Case Report of Surgical Nephrectomy, Diagnostic Imaging, and Long-Term Outcomes.
CureusPostnatal outcomes and surgical management of prenatally detected unilateral congenital anomalies of the kidney and urinary tract.
Minerva obstetrics and gynecologyPrenatal diagnosis of fetuses with renal abnormalities: a retrospective analysis of 329 Chinese cases.
Orphanet journal of rare diseasesCase Report: Diagnostic overlap of OHVIRA syndrome and Gartner duct cyst: challenges in imaging and management.
Frontiers in pediatricsZinner's syndrome in two young middle-aged men: a case report and review of the literature.
BMC urologyUnilateral Inguinal Swelling in a Young Female: An Unusual Presentation of MURCS.
Journal of human reproductive sciencesPilot Study of Using Machine Learning to Detect Atherosclerotic Renal Artery Stenosis From Spectral Doppler Waveforms.
Kidney international reportsA Rare First Presentation of Unilateral Fibromuscular Dysplasia Resulting in Renal Infarct.
CureusDoes unilateral reflux have a protective effect in posterior urethral valve patients?
Pediatric surgery internationalEctopic ureter presenting as a scrotal fistula associated with unilateral atrophic kidney: a rare case report.
Annals of medicine and surgery (2012)[Perioperative management of cochlear implantation and analysis on the influencing factors of efficacy in patients diagnosed as hereditary syndromic hearing loss].
Zhonghua er bi yan hou tou jing wai ke za zhi = Chinese journal of otorhinolaryngology head and neck surgeryFetal bilateral hyperechogenic kidneys: Prenatal progression and long-term postnatal outcome.
Early human developmentA rare variant of zinner syndrome with ejaculatory duct cyst: case report and challenges in diagnosis and management.
BMC urologySupporting Infants with Multicystic Dysplastic Kidney Disease: A Comprehensive Approach.
Neonatal network : NNDistinguishing Features of Childhood Renal Dysplasia.
Klinische PadiatrieAssessment of ureteric jets as a supportive diagnostic modality for unilateral pelvi-ureteric junction obstruction and its utility in follow-up: A pilot study.
Journal of pediatric urologyPediatric Zinner syndrome variants: Case series with newer insights into pathogenesis in early childhood.
Journal of pediatric urologyZinner syndrome with contralateral hydronephrosis: A rare congenital condition.
Urology case reportsZinner Syndrome in Young Adult Males: A Case Series and Literature Review.
CureusRenal artery occlusion in a young woman - a tale of mysterious thrombosis.
VascularA novel homozygous splice site variant in ARL2BP causes a syndromic autosomal recessive rod-cone dystrophy with situs inversus, asthenozoospermia, unilateral renal agenesis and microcysts.
BMC medical genomicsA novel heterozygous variant of the SALL1 gene with atypical Townes-Brocks syndrome phenotypes in Chinese family.
Nephrology (Carlton, Vic.)Early post-operative outcomes of robot-assisted pyeloplasty in patients with unilateral ureteropelvic junction obstruction.
International urology and nephrologyMulticystic renal dysplasia, a histomorphological spectrum: Seven years experience from a tertiary care hospital.
Indian journal of pathology & microbiologyA renal aplasia case mimicking radiologically as unilateral renal agenesis in a child with spina bifida, atresia ani and unilateral undescended testis: a case report.
Journal of medical case reportsCase report: A novel mutation in the EYA1 gene in a child with branchiootic syndrome with secretory otitis media and bilateral vestibular hypofunction.
Frontiers in geneticsAnalysis of the Efficacy of Elastography in Comparison with Dynamic Renal Nuclear Scintigraphy in the Evaluation of Unilateral Pelvi-Ureteric Junction Obstruction.
Journal of pediatric surgeryTroubleshooting Tips for Diagnosing Complex Fetal Genitourinary Malformations.
Radiographics : a review publication of the Radiological Society of North America, IncClinicopathological and genetic features of Zinner's syndrome: two case reports and review of the literature.
Frontiers in urologyPosterior Urethral Valves, Unilateral Vesicoureteral Reflux, and Renal Dysplasia (VURD) Syndrome: Long-Term Longitudinal Evaluation of the Kidney Function.
International journal of environmental research and public healthFibromuscular Dysplasia Presenting as Acute Unilateral Renal Infarction: A Case Report and Review of Two Diseases.
CureusManagement dilemma in pelvi-ureteric junction obstruction: is transit time the answer?
Pediatric surgery internationalEctopic Ureter-A Retrospective Analysis, Symptom and Treatment.
Archivos espanoles de urologiaCraniosynostosis, inner ear, and renal anomalies in a child with complete loss of SPRY1 (sprouty homolog 1) function.
Journal of medical geneticsPop-off mechanisms as protective factors against chronic renal disease in children with posterior urethral valves.
Cirugia pediatrica : organo oficial de la Sociedad Espanola de Cirugia PediatricaUnilateral macrocystic dysplasia and contralateral agenesis in a monoamniotic twin.
Ceska gynekologieANOS1 variants in a large cohort of Chinese patients with congenital hypogonadotropic hypogonadism.
Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciencesUtility of trio-based prenatal exome sequencing incorporating splice-site and mitochondrial genome assessment in pregnancies with fetal ultrasound anomalies: prospective cohort study.
Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and GynecologyCystic dysplasia of the rete testis associated with ipsilateral renal agenesis: A case report.
Radiology case reportsRenal Artery Variations in Patients With Mild-to-Moderate Hypertension From the RADIANCE-HTN SOLO Trial.
Cardiovascular revascularization medicine : including molecular interventionsRenal Artery Reconstruction for Refractory Hypertension Caused by Congenital Renal Artery Deficiency.
Annals of vascular surgeryThe obstructive index in antenatal unilateral pelviureteric junction obstruction: A novel predictor of the failure of conservative management.
Pediatrics international : official journal of the Japan Pediatric SocietyA rare case of symptomatic hydrometrocolpos in a 5y old female.
Urology case reportsAssociation of prenatal renal ultrasound abnormalities with pathogenic copy number variants in a large Chinese cohort.
Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and GynecologyUnilateral Urogenital Disontogeny in a Dog.
Case reports in veterinary medicineClinical characteristics and long-term outcomes of endovascular treatment of renal artery fibromuscular dysplasia with branch lesions.
Pediatric nephrology (Berlin, Germany)Fibromuscular dysplasia with unilateral renal agenesis.
BMJ case reportsNeonatal Respiratory Distress and Airway Emergency: Report of Two Cases.
Children (Basel, Switzerland)Clinical characteristics of 1,055 Chinese patients with Mayer-Rokitansky-Küster-Hauser syndrome: a nationwide multicentric study.
Fertility and sterilityPrenatal diagnosis of intestinal nonrotation using magnetic resonance imaging: Is it possible?
Pediatric radiologyDiagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021.
American journal of kidney diseases : the official journal of the National Kidney FoundationPrenatal Diagnosis and Findings in Ureteropelvic Junction Type Hydronephrosis.
Frontiers in pediatricsTP63-mutation as a cause of prenatal lethal multicystic dysplastic kidneys.
Molecular genetics & genomic medicineCongenital Unilateral Renal Aplasia in a Cynomolgus Monkey (Macaca fascicularis) With Investigation Into Potential Pathogenesis.
Toxicologic pathologyUltrasound Imaging of Renal Cysts in Children.
Journal of ultrasound in medicine : official journal of the American Institute of Ultrasound in Medicine[Kidney Cysts and Cystic Nephropathies in Children - A Consensus Guideline by 10 German Medical Societies].
Klinische PadiatrieSirenomelia (Mermaid Syndrome): A Case Report.
Turk patoloji dergisiIdentification of two novel mutations in three Chinese families with Kallmann syndrome using whole exome sequencing.
AndrologiaA simple, refined approach to diagnosing renovascular hypertension in children: A 10-year study.
Pediatrics international : official journal of the Japan Pediatric SocietyEfficacy of Antihypertensive Therapy in a Child with Unilateral Focal Fibromuscular Dysplasia of the Renal Artery: A Case Study and Review of Literature.
Medicines (Basel, Switzerland)Isolated Renal Artery Dissection: A Systematic Review of Case Reports.
CureusDiagnosis of fetal non-chromosomal abnormalities on routine ultrasound examination at 11-13 weeks' gestation.
Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and GynecologyA Case of Mayer-Rokitansky-Küster-Hauser Syndrome with a Fused Pancake-shaped Pelvic Kidney.
Advanced biomedical researchLong-term growth in children with posterior urethral valves.
Journal of pediatric urologyHNF1B nephropathy has a slow-progressive phenotype in childhood-with the exception of very early onset cases: results of the German Multicenter HNF1B Childhood Registry.
Pediatric nephrology (Berlin, Germany)Successful endovascular treatment of chronic renal artery occlusion: a preliminary retrospective case series including 15 patients.
International urology and nephrologyUnilateral segmental dysplasia of the vas deferens.
The Canadian journal of urologyIncreased Sylvian fissure angle as early sonographic sign of malformation of cortical development.
Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and GynecologyDifferences in renal hemodynamics and renin secretion between patients with unifocal and multifocal fibromuscular dysplasia.
Journal of hypertensionThe distinct optic disk and peripapillary appearance in Donnai-Barrow syndrome.
Ophthalmic geneticsEsophageal atresia with distal fistula - unusual case series. Considerations related to epidemiological aspects, malformative associations, and prenatal diagnosis.
Romanian journal of morphology and embryology = Revue roumaine de morphologie et embryologieInvolvement of the bone morphogenic protein/SMAD signaling pathway in the etiology of congenital anomalies of the kidney and urinary tract accompanied by cryptorchidism.
BMC urologyUrinary carbohydrate antigen 19-9/creatinine ratio: A non-invasive marker for follow-up of unilateral ureteropelvic junction obstruction in children.
Journal of pediatric urologyAnalysis of renal blood flow and renal volume in normal fetuses and in fetuses with a solitary functioning kidney.
Prenatal diagnosisPredictive value of cortical transit time on MAG3 for surgery in antenatally detected unilateral hydronephrosis caused by ureteropelvic junction stenosis.
Journal of pediatric urologyUnilateral multicystic renal dysplasia: Prenatal diagnosis on ultrasound.
Saudi journal of kidney diseases and transplantation : an official publication of the Saudi Center for Organ Transplantation, Saudi ArabiaHigh Prevalence of Multiple Arterial Bed Lesions in Patients With Fibromuscular Dysplasia: The ARCADIA Registry (Assessment of Renal and Cervical Artery Dysplasia).
Hypertension (Dallas, Tex. : 1979)Large Multicystic Dysplastic Kidney Mimicking a Large Cystic Renal Neoplasm.
Journal of clinical and diagnostic research : JCDRIs it Always Necessary to Treat an Asymptomatic Hydronephrosis Due to Ureteropelvic Junction Obstruction?
Indian journal of pediatricsProximal ureteral atresia, a rare congenital anomaly-incidental finding: a case report.
Translational pediatricsPathophysiological differences between multifocal fibromuscular dysplasia and atherosclerotic renal artery stenosis.
Journal of hypertensionEpididymis-like Tubules in Adult Renal Hypodysplasia: Immunohistochemical Features Indicate a Mesonephric Origin.
International journal of surgical pathologyDiagnostic imaging and cataloguing of female genital malformations.
Insights into imagingFetal anomalies associated with HNF1B mutations: report of 20 autopsy cases.
Prenatal diagnosisSpondyloepimetaphyseal dysplasia with joint laxity (Beighton type): A unique South African disorder.
South African medical journal = Suid-Afrikaanse tydskrif vir geneeskundeACE serum level and I/D gene polymorphism in children with obstructive uropathies and other congenital anomalies of the kidney and urinary tract.
Nephrology (Carlton, Vic.)Renal hemodynamics and renin-angiotensin system activity in humans with multifocal renal artery fibromuscular dysplasia.
Journal of hypertensionUnilateral congenital giant megaureter with renal dysplasia compressing contralateral ureter and causing bilateral hydronephrosis: a case report and literature review.
BMC urologyOutcome after prenatal diagnosis of congenital anomalies of the kidney and urinary tract.
European journal of pediatricsRenal Damage Frequency in Patients with Solitary Kidney and Factors That Affect Progression.
International journal of nephrologyAssociated extrarenal vascular diseases may complicate the treatment and outcome of renovascular hypertension.
Acta paediatrica (Oslo, Norway : 1992)The MURCS Association: Mullerian Duct Aplasia, Renal Hypoplasia and Cervicothoracic Somite Dysplasia - A Case Report.
Mymensingh medical journal : MMJ[Efficiency evaluation of diuretic renography in the operative or conservative treatments of unilateral ureteropelvic junction obstruction patients].
Beijing da xue xue bao. Yi xue ban = Journal of Peking University. Health sciencesURETERIC ANGIOMYOLIPOMA CAUSING UNILATERAL PELVI-URETERIC JUNCTION OBSTRUCTION.
Journal of Ayub Medical College, Abbottabad : JAMCMeckel-Gruber Syndrome with unilateral renal agenesis.
Journal of the College of Physicians and Surgeons--Pakistan : JCPSPAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Displasia renal, unilateral.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Displasia renal, unilateral
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Unilateral Multicystic Dysplastic Kidney in a Fetus Associated With Parental Genetic and Environmental Risk Factors: A Case Report.
- Prevalence, clinical profile, and associated anomalies with women with Mayer-Rokitansky-Küster-Hauser syndrome in a tertiary care center: A cross-sectional study.International journal of gynaecology and obstetrics: the official organ of the International Federation of Gynaecology and Obstetrics· 2026· PMID 41636313mais citado
- Prenatal diagnosis of fetuses with renal abnormalities: a retrospective analysis of 329 Chinese cases.
- Case Report: Diagnostic overlap of OHVIRA syndrome and Gartner duct cyst: challenges in imaging and management.
- Unilateral Inguinal Swelling in a Young Female: An Unusual Presentation of MURCS.
- Transient neonatal diabetes mellitus as an early diagnostic clue to HNF1B-related disease - two case reports and a literature review.
- Mayer-Rokitansky-Küster-Hauser Syndrome Associated With Diabetes Mellitus and Renal Anomalies in an Adolescent Girl: A Rare Case Report.
- Adult Diagnosis of Solitary Kidney and Renal Dysplasia in a Male Born Prematurely as a Twin: A Case Report.
- Meckel-Gruber syndrome: a rare and fatal congenital disorder (case report).
- Aorto-Renal Dysplasia in Childhood: The Overlap of Neurofibromatosis Type 1 and Pediatric Renovascular Hypertension.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:93172(Orphanet)
- MONDO:0019644(MONDO)
- GARD:19177(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55788766(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Displasia renal, unilateral
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata