Síndrome de Beckwith-Wiedemann causada por dissomia uniparental paterna do cromossomo 11, resultando em crescimento excessivo e anomalias congênitas.
Introdução
O que você precisa saber de cara
Visão geral
A Dissomia uniparental de origem paterna, cromossomo 11 é uma condição genética rara que faz parte do espectro da síndrome de Beckwith-Wiedemann. Nesta condição, a pessoa herda duas cópias do cromossomo 11 do pai, em vez de uma cópia de cada genitor. Isso leva a um desequilíbrio na expressão de genes importantes para o crescimento e desenvolvimento, resultando em características como crescimento excessivo (macrossomia), aumento de alguns órgãos e risco aumentado para tumores na infância.[1]
Sinais e sintomas
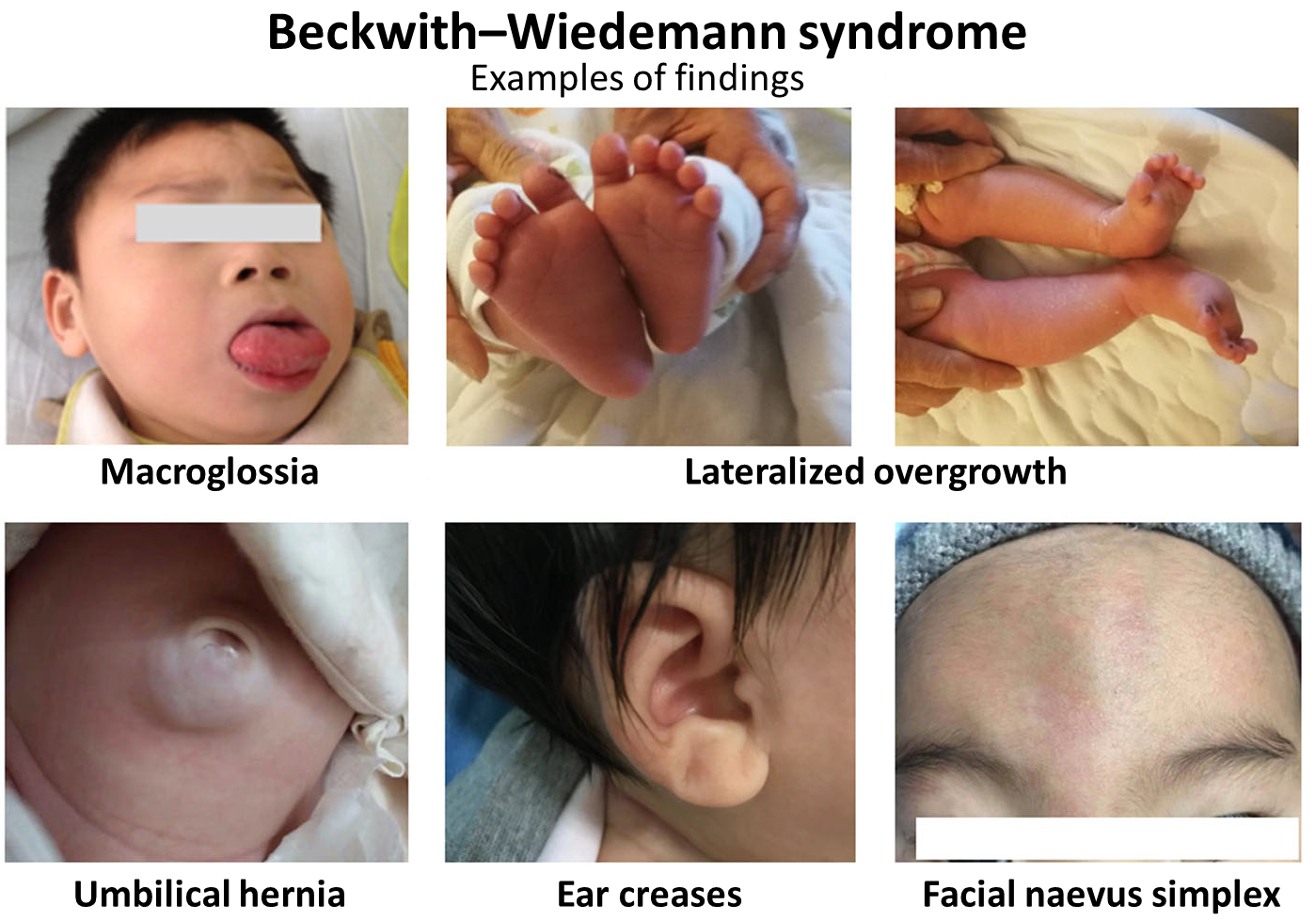
Os sinais e sintomas mais comuns incluem crescimento excessivo antes e depois do nascimento, aumento da língua (macroglossia), defeitos na parede abdominal (como onfalocele ou hérnia umbilical), pregas nas orelhas e risco aumentado para tumores embrionários, como tumor de Wilms e hepatoblastoma. A gravidade dos sintomas pode variar amplamente entre os indivíduos.[1]
Causas genéticas
A condição é causada pela dissomia uniparental paterna do cromossomo 11, ou seja, a presença de duas cópias do cromossomo 11 herdadas exclusivamente do pai. Isso altera a expressão de genes localizados em regiões específicas do cromossomo 11, especialmente aqueles sujeitos ao imprinting genômico (mecanismo que faz com que certos genes sejam expressos apenas de um dos pais). O gene específico envolvido não foi identificado nos dados disponíveis.[1][3]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sinais característicos e confirmado por testes genéticos. Os exames disponíveis no SUS incluem: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; e sequenciamento completo do exoma (WES). A dosagem de alfa-fetoproteína pode ser utilizada para monitoramento de tumores. O código CID-10 associado é Q87.3.[1]
Tratamento e manejo
O manejo é multidisciplinar e inclui acompanhamento regular para detecção precoce de tumores (como ultrassonografia abdominal e dosagem de alfa-fetoproteína), tratamento de macroglossia quando necessário, correção cirúrgica de defeitos da parede abdominal e suporte para atrasos no desenvolvimento. No SUS, há cobertura parcial para procedimentos como atendimento em reabilitação para doenças raras. Não há medicamentos específicos aprovados para a condição; o tratamento é sintomático e de suporte.[1]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade dos sintomas e a presença de tumores. Com acompanhamento médico adequado e detecção precoce de complicações, muitas pessoas com a condição têm boa qualidade de vida. O risco de tumores diminui após a infância, e a maioria dos adultos leva uma vida saudável, embora possam necessitar de acompanhamento contínuo.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Síndrome de Beckwith-Wiedemann causada por dissomia uniparental paterna do cromossomo 11, resultando em crescimento excessivo e anomalias congênitas.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Dissomia uniparental de origem paterna, cromossomo 11 é uma condição genética rara que faz parte do espectro da síndrome de Beckwith-Wiedemann. Nesta condição, a pessoa herda duas cópias do cromossomo 11 do pai, em vez de uma cópia de cada genitor. Isso leva a um desequilíbrio na expressão de genes importantes para o crescimento e desenvolvimento, resultando em características como crescimento excessivo (macrossomia), aumento de alguns órgãos e risco aumentado para tumores na infância.[1]
Sinais e sintomas
Os sinais e sintomas mais comuns incluem crescimento excessivo antes e depois do nascimento, aumento da língua (macroglossia), defeitos na parede abdominal (como onfalocele ou hérnia umbilical), pregas nas orelhas e risco aumentado para tumores embrionários, como tumor de Wilms e hepatoblastoma. A gravidade dos sintomas pode variar amplamente entre os indivíduos.[1]
Causas genéticas
A condição é causada pela dissomia uniparental paterna do cromossomo 11, ou seja, a presença de duas cópias do cromossomo 11 herdadas exclusivamente do pai. Isso altera a expressão de genes localizados em regiões específicas do cromossomo 11, especialmente aqueles sujeitos ao imprinting genômico (mecanismo que faz com que certos genes sejam expressos apenas de um dos pais). O gene específico envolvido não foi identificado nos dados disponíveis.[1][3]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sinais característicos e confirmado por testes genéticos. Os exames disponíveis no SUS incluem: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; e sequenciamento completo do exoma (WES). A dosagem de alfa-fetoproteína pode ser utilizada para monitoramento de tumores. O código CID-10 associado é Q87.3.[1]
Tratamento e manejo
O manejo é multidisciplinar e inclui acompanhamento regular para detecção precoce de tumores (como ultrassonografia abdominal e dosagem de alfa-fetoproteína), tratamento de macroglossia quando necessário, correção cirúrgica de defeitos da parede abdominal e suporte para atrasos no desenvolvimento. No SUS, há cobertura parcial para procedimentos como atendimento em reabilitação para doenças raras. Não há medicamentos específicos aprovados para a condição; o tratamento é sintomático e de suporte.[1]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade dos sintomas e a presença de tumores. Com acompanhamento médico adequado e detecção precoce de complicações, muitas pessoas com a condição têm boa qualidade de vida. O risco de tumores diminui após a infância, e a maioria dos adultos leva uma vida saudável, embora possam necessitar de acompanhamento contínuo.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Dissomia uniparental de origem paterna, cromossomo 11
Centros de Referência SUS
24 centros habilitados pelo SUS para Dissomia uniparental de origem paterna, cromossomo 11
Centros para Dissomia uniparental de origem paterna, cromossomo 11
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Comparison of Minimally Invasive Surfactant Therapy and Intubation-surfactant Administration-extubation in Premature Neonates with Respiratory Distress Syndrome.
Beckwith-Wiedemann syndrome (BWS) is a rare genetic and cancer-predisposing disorder characterized by variable clinical and molecular abnormalities. It is considered a spectrum ranging from classical BWS to isolated hemihyperplasia (IHH). This study sought to characterize Omani patients with BWS and IHH clinically and molecularly, evaluate their surveillance results, and assess the tumor's prevalence in the cohort. Nine patients with BWS were retrospectively recruited to the study by searching the medical records of Sultan Qaboos University Hospital between January 2012 and December 2022. Demographics, clinical features, molecular findings, and surveillance test results, including abdominal ultrasound and alpha-fetoprotein, were extracted from the hospital information system and systematically analyzed. Nine patients diagnosed with Beckwith-Wiedemann syndrome were studied, comprising four BWS cases and five IHH cases. Macroglossia was the predominant clinical feature among BWS patients, whereas lateralized overgrowth was consistently observed in IHH patients. All BWS patients tested positive for methylation anomalies: two exhibited loss of methylation at imprinting control 2 (22.2%), one had paternal uniparental disomy of chromosome 11 (11.1%), and another showed a gain of methylation at imprinting control 1 (11.1%). Throughout the surveillance period, none of the patients showed elevated alpha-fetoprotein levels or developed tumors. This is the first study to examine a cohort of patients with BW spectrum in Oman. It reveals comparable clinical and molecular characteristics to the previously reported BWS patients, yet no tumors were detected in this cohort.
Beckwith-Wiedemann syndrome with juvenile fibrous nodules and lobular breast tumors: a case report and review of the literature.
Beckwith-Wiedemann syndrome (BWS) is a genomic imprinting disorder caused by diverse genetic and/or epigenetic disorders of chromosome 11p15.5. BWS presents with a variety of clinical features, including overgrowth and an increased risk of embryonal tumors. Notably however, reports of patients with BWS and breast tumors are rare, and the association between these conditions is still unclear. Insulin-like growth factor-2 (IGF2) expression is known to be associated with the development of various cancers, including breast cancer, and patients with BWS with specific subtypes of molecular defects are known to show characteristic clinical features and IGF2 overexpression. A 17-year-old girl who had been diagnosed with BWS based on an umbilical hernia, hyperinsulinemia, and left hemihypertrophy at birth, visited our department with a gradually swelling left breast. Her left breast was markedly larger than her right breast on visual examination. Imaging examinations showed two tumors measuring about 10 cm each in the left breast, and she was diagnosed with juvenile fibroadenoma following core needle biopsy. The two breast tumors were removed surgically and the patient remained alive with no recurrence. The final diagnosis was juvenile fibroadenoma without malignant findings. Immunohistochemical staining using IGF2 antibody revealed overexpression of IGF2 in the cytoplasm of ductal epithelial cells. Because of her clinical features and IGF2 overexpression, molecular defects of 11p15.5 including a possible genetic background of paternal uniparental disomy of chromosome 11 or hypermethylation of imprinting center 1 was suspected. In this case, overexpression of IGF2 suggested a possible relationship between BWS and breast tumors. Moreover, the characteristic clinical features and IGF2 staining predicted the subtype of 11p15.5 molecular defects in this patient.
Placental Mesenchymal Dysplasia and Beckwith-Wiedemann Syndrome.
Placental mesenchymal dysplasia (PMD) is characterized by placentomegaly, aneurysmally dilated chorionic plate vessels, thrombosis of the dilated vessels, and large grapelike vesicles, and is often mistaken for partial or complete hydatidiform mole with a coexisting normal fetus. Androgenetic/biparental mosaicism (ABM) has been found in many PMD cases. Beckwith-Wiedemann syndrome (BWS) is an imprinting disorder with complex and diverse phenotypes and an increased risk of developing embryonal tumors. There are five major causative alterations: loss of methylation of imprinting control region 2 (KCNQ1OT1:TSS-DMR) (ICR2-LOM), gain of methylation at ICR1 (H19/IGF2:IG-DMR) (ICR1-GOM), paternal uniparental disomy of 11 (pUPD11), loss-of-function variants of the CDKN1C gene, and paternal duplication of 11p15. Additional minor alterations include genetic variants within ICR1, paternal uniparental diploidy/biparental diploidy mosaicism (PUDM, also called ABM), and genetic variants of KCNQ1. ABM (PUDM) is found in both conditions, and approximately 20% of fetuses from PMD cases are BWS and vice versa, suggesting a molecular link. PMD and BWS share some molecular characteristics in some cases, but not in others. These findings raise questions concerning the timing of the occurrence of the molecularly abnormal cells during the postfertilization period and the effects of these abnormalities on cell fates after implantation.
Further understanding of paternal uniparental disomy in Beckwith-Wiedemann syndrome.
Paternal uniparental disomy of chromosome 11 (upd(11)pat) accounts for up to 20% of molecularly confirmed Beckwith-Wiedemann spectrum (BWSp) cases. It belongs to the BWSp subgroup with the second highest tumor risk, and therefore needs particular awareness in research, diagnostics and clinical management. We overview the contribution of paternal (mosaic) uniparental disomy of chromosome 11 (UPD, upd(11)pat) and mosaic paternal uniparental diploidy in patients with Beckwith-Wiedemann features. The review comprises the current knowledge on their formation and their molecular and clinical consequences. Accordingly, the consequences for diagnostic testing and clinical monitoring are compiled. The necessity to diagnostically identify and thus discriminate genome-wide paternal uniparental disomy, and upd(11)pat becomes obvious, due to the differences in the clinical course, disease prognosis, and treatment. In particular, monitoring of tumor development by liquid biopsy might be a promising option in the future. From the research point of view, it should be addressed why 11p is prone to mitotic recombination and thus also provide to the role of upd(11) as second hit in tumorigenesis.
Adrenocortical Tumors in Children With Constitutive Chromosome 11p15 Paternal Uniparental Disomy: Implications for Diagnosis and Treatment.
Pediatric adrenocortical tumors (ACTs) are rare and heterogeneous. Approximately 50% of children with ACT carry a germline TP53 variant; however, the genetic underpinning of remaining cases has not been elucidated. In patients having germline TP53 variants, loss of maternal chromosome 11 and duplication of the paternal copy [paternal uniparental disomy, (UPD)] occurs early in tumorigenesis and explains the overexpression of IGF2, the hallmark of pediatric ACT. Beckwith-Wiedemann syndrome (BWS) is also associated with overexpression of IGF2 due to disruption of the 11p15 loci, including segmental UPD. Here, we report six children with ACT with wild type TP53 and germline paternal 11p15 UPD. Median age of five girls and one boy was 3.2 years (range 0.5-11 years). Two patients met the criteria for BWS before diagnosis of ACT. However, ACT was the first and only manifestation of paternal 11p15 UPD in four children. Tumor weight ranged from 21.5 g to 550 g. Despite poor prognostic features at presentation, such as pulmonary metastasis, bilateral adrenal involvement, and large tumors, all patients are alive 8-21 years after cancer diagnosis. Our observations suggest that children with ACT and wild type TP53, irrespective of their age, should be screened for germline abnormalities in chromosome 11p15.
Publicações recentes
Further understanding of paternal uniparental disomy in Beckwith-Wiedemann syndrome.
Adrenocortical Tumors in Children With Constitutive Chromosome 11p15 Paternal Uniparental Disomy: Implications for Diagnosis and Treatment.
Pediatric Cushing syndrome: An early sign of an underling cancer predisposition syndrome.
Mosaic Segmental and Whole-Chromosome Upd(11)mat in Silver-Russell Syndrome.
Diffuse infantile hepatic hemangiomas in a patient with Beckwith-Wiedemann syndrome: A new association?
📚 EuropePMCmostrando 11
Comparison of Minimally Invasive Surfactant Therapy and Intubation-surfactant Administration-extubation in Premature Neonates with Respiratory Distress Syndrome.
Oman medical journalBeckwith-Wiedemann syndrome with juvenile fibrous nodules and lobular breast tumors: a case report and review of the literature.
Surgical case reportsPlacental Mesenchymal Dysplasia and Beckwith-Wiedemann Syndrome.
CancersFurther understanding of paternal uniparental disomy in Beckwith-Wiedemann syndrome.
Expert review of endocrinology & metabolismAdrenocortical Tumors in Children With Constitutive Chromosome 11p15 Paternal Uniparental Disomy: Implications for Diagnosis and Treatment.
Frontiers in endocrinologyBeckwith-Wiedemann syndrome: Clinical, histopathological and molecular study of two Tunisian patients and review of literature.
Molecular genetics & genomic medicineMosaic Segmental and Whole-Chromosome Upd(11)mat in Silver-Russell Syndrome.
GenesClinical, Genetic, and Prognostic Features of Adrenocortical Tumors in Children: A 10-Year Single-Center Experience.
Frontiers in oncologyAndrogenetic chimerism as an etiology for Beckwith-Wiedemann syndrome: diagnosis and management.
Genetics in medicine : official journal of the American College of Medical GeneticsA multi-method approach to the molecular diagnosis of overt and borderline 11p15.5 defects underlying Silver-Russell and Beckwith-Wiedemann syndromes.
Clinical epigeneticsIdentification of consensus motifs associated with mitotic recombination and clinical characteristics in patients with paternal uniparental isodisomy of chromosome 11.
Human molecular geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Dissomia uniparental de origem paterna, cromossomo 11.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Dissomia uniparental de origem paterna, cromossomo 11
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Comparison of Minimally Invasive Surfactant Therapy and Intubation-surfactant Administration-extubation in Premature Neonates with Respiratory Distress Syndrome.
- Beckwith-Wiedemann syndrome with juvenile fibrous nodules and lobular breast tumors: a case report and review of the literature.
- Placental Mesenchymal Dysplasia and Beckwith-Wiedemann Syndrome.
- Further understanding of paternal uniparental disomy in Beckwith-Wiedemann syndrome.
- Adrenocortical Tumors in Children With Constitutive Chromosome 11p15 Paternal Uniparental Disomy: Implications for Diagnosis and Treatment.
- Pediatric Cushing syndrome: An early sign of an underling cancer predisposition syndrome.
- Mosaic Segmental and Whole-Chromosome Upd(11)mat in Silver-Russell Syndrome.
- Diffuse infantile hepatic hemangiomas in a patient with Beckwith-Wiedemann syndrome: A new association?
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:96193(Orphanet)
- MONDO:0019923(MONDO)
- GARD:19342(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q55788998(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Dissomia uniparental de origem paterna, cromossomo 11
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub