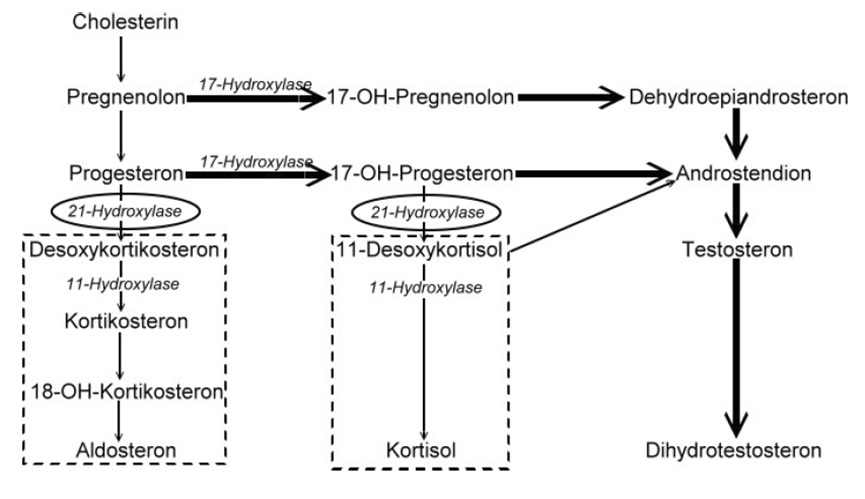
A forma virilizante simples da hiperplasia adrenal congênita clássica (HAC clássica), que é causada pela deficiência da enzima 21-hidroxilase, caracteriza-se por genitália ambígua (ou seja, os órgãos sexuais externos não são claramente masculinos nem femininos) e masculinização da genitália externa em meninas, baixos níveis do hormônio cortisol e pseudopuberdade precoce (sinais de puberdade que surgem antes do tempo normal, mas não são uma puberdade completa), sem perda excessiva de sal pelo organismo.
Introdução
O que você precisa saber de cara
A forma virilizante simples da hiperplasia adrenal congênita clássica (HAC clássica), que é causada pela deficiência da enzima 21-hidroxilase, caracteriza-se por genitália ambígua (ou seja, os órgãos sexuais externos não são claramente masculinos nem femininos) e masculinização da genitália externa em meninas, baixos níveis do hormônio cortisol e pseudopuberdade precoce (sinais de puberdade que surgem antes do tempo normal, mas não são uma puberdade completa), sem perda excessiva de sal pelo organismo.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Linha do tempo da pesquisa
Triagem neonatal (Teste do Pezinho)
A triagem neonatal permite diagnóstico precoce e início imediato do tratamento.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
A cytochrome P450 monooxygenase that plays a major role in adrenal steroidogenesis. Catalyzes the hydroxylation at C-21 of progesterone and 17alpha-hydroxyprogesterone to respectively form 11-deoxycorticosterone and 11-deoxycortisol, intermediate metabolites in the biosynthetic pathway of mineralocorticoids and glucocorticoids (PubMed:10602386, PubMed:16984992, PubMed:22014889, PubMed:25855791, PubMed:27721825). Mechanistically, uses molecular oxygen inserting one oxygen atom into a substrate, a
Endoplasmic reticulum membraneMicrosome membrane
Adrenal hyperplasia 3
A form of congenital adrenal hyperplasia, a common recessive disease due to defective synthesis of cortisol. Congenital adrenal hyperplasia is characterized by androgen excess leading to ambiguous genitalia in affected females, rapid somatic growth during childhood in both sexes with premature closure of the epiphyses and short adult stature. Four clinical types: 'salt wasting' (SW, the most severe type), 'simple virilizing' (SV, less severely affected patients), with normal aldosterone biosynthesis, 'non-classic form' or late-onset (NC or LOAH) and 'cryptic' (asymptomatic).
Medicamentos aprovados (FDA)
1 medicamento encontrado nos registros da FDA americana.
Variantes genéticas (ClinVar)
206 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
4 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Hiperplasia suprarrenal congênita clássica por deficiência de 21-hidroxilase, forma virilizante simples
Centros de Referência SUS
24 centros habilitados pelo SUS para Hiperplasia suprarrenal congênita clássica por deficiência de 21-hidroxilase, forma virilizante simples
Centros para Hiperplasia suprarrenal congênita clássica por deficiência de 21-hidroxilase, forma virilizante simples
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Genotype-Phenotype Correlation in Children With Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency Using Next Generation Sequencing.
Although there a well-known correlation in genotype and phenotype, patients with 21-OHD caused by severe pathogenic variants have better correlation, whereas inconsistencies are more common in the presence of milder variants. This study aimed to evaluate CYP21A2 genotyping and reveal the genotype-phenotype correlation in children diagnosed with 21-OHD in the South-eastern Anatolia region, where ethnic diversity and consanguineous marriage rates are high. The patients were divided into three groups: salt wasting (SW), simple virilizing (SV) and non-classical (NC). Pathogenic variants of the CYP21A2 gene were classified into six groups based on predicted 21-hydroxylase activity: null-A-B-C-D-E. CYP21A2 genotyping was performed by sequence-specific primer and sequenced with next generation sequencing (NGS), and the expected phenotypes were compared to the observed phenotypes. The overall genotype-phenotype concordance was found to be 73.1% (68/93). The expected concordance with the SW form of the null (n = 12) and A (n = 51) groups is 91.6% and 88.2%, respectively. While the percentage of the expected clinical form of SV in patients in group B (n = 5) was 80%, the concordance for the expected clinical form of NC for group C (n = 25) was not strong enough (32%). This study demonstrates that children with 21-hydroxylase deficiency show a good correlation between severe pathogenic variants and predicted clinical phenotypes; however, the correlation is not strong enough between milder variants. The discrepancies could have resulted from the complex characteristics of 21-OHD genotyping and the limitations of using NGS alone without integrating with other comprehensive methods.
Nationwide carrier screening for congenital adrenal hyperplasia: integrated approach of CYP21A2 pathogenic variant genotyping and comprehensive large gene deletion analysis.
Congenital Adrenal Hyperplasia (CAH) due to 21-hydroxylase deficiency (21-OHD CAH) is an autosomal recessive disorder resulting from pathogenic variants in the CYP21A2 gene. The disorder exhibits variable clinical severity, with the classical form manifesting as salt-wasting crisis in neonates, while inducing ambiguous genitalia in females and precocious puberty in males through simple virilization. Identifying at-risk couples during the preconception stage holds significance for optimizing reproductive choices. This study included 204 unrelated preconception individuals undergoing carrier screening. A robust molecular approach was devised for rapid detection of nine prevalent CYP21A2 pathogenic variants, utilizing Amplification-Refractory Mutation System (ARMS) PCR and mass spectrometry (MS) genotyping. Complementary quantitative real-time PCR (qPCR) and PCR-based Restriction Fragment Length Polymorphism (PCR-based RFLP) assays were employed for comprehensive gene deletion analysis. The concordance of pathogenic variant detection between ARMS-PCR and MS, as well as the consistency observed in molecular insights from qPCR and PCR-based RFLP, fortified the accuracy of our methodologies. Our combined method could detect common pathogenic variants and large gene deletions with high concordance between ARMS-PCR, MS genotyping, qPCR, and PCR-based RFLP assays. Remarkably, two carriers exhibited significant large-scale deletions, while another manifested a carrier state due to minor-scale gene conversion. The estimated carrier frequency in our cohort using these methods was approximately 1 in 65 individuals. The methods used for 21-OHD CAH carrier screening offer a reliable, swift, and cost-effective approach for detecting common pathogenic variants and large deletions. Despite some limitations, such as the inability to detect all rare mutations, the techniques provide a practical solution for carrier screening, with an estimated carrier frequency of 1 in 65 in our study population. These findings support the potential adoption of these methods in national carrier screening programs, offering a practical balance between efficiency and affordability.
Genetics and Pathophysiology of Classic Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency.
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disease that manifests clinically in varying forms depending on the degree of enzyme deficiency. CAH is most commonly caused by 21-hydroxylase deficiency (21OHD) due to mutations in the CYP21A2 gene. Whereas there is a spectrum of disease severity, 21OHD is generally categorized into 3 forms. The classic form encompasses salt-wasting and simple virilizing CAH and the least affected form is termed nonclassic CAH. The classic form of 21OHD occurs in ∼1 in 16 000 births with the most severe salt-wasting cases presenting in the neonatal period with cortisol and aldosterone deficiencies and virilization of external female genitalia. Cortisol deficiency removes normal feedback on the hypothalamic-pituitary-adrenal axis leading to elevations in ACTH and adrenal androgen levels, which often accelerate skeletal maturation, leading to premature epiphyseal growth plate closure. Additionally, supraphysiologic doses of glucocorticoids are necessary to suppress androgen levels, adversely affecting final adult height. This paper highlights a brief history of 21OHD and provides an overview of the genetic basis and pathophysiology of 21OHD.
Clinical, Biochemical and Molecular Characteristics of Congenital Adrenal Hyperplasia Due to 21-hydroxylase Deficiency.
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disease caused by the deficiency of one of the enzymes involved in cortisol synthesis. Between 90% and 99% of cases of CAH are caused by 21-hydroxylase deficiency (21-OHD) caused by mutations in CYP21A2. Although 21-OHD has been historically divided into classical and non-classical forms, it is now thought to show a continuous phenotype. In the classical form, the external genitalia in females becomes virilized to varying degrees. If the disease is not recognized, salt wasting crises in the classical form may threaten life in neonates. Children experience accelerated somatic growth, increased bone age, and premature pubic hair in the simple virilizing form of classical 21-OHD. Female adolescents may present with severe acne, hirsutism, androgenic alopecia, menstrual irregularity or primary amenorrhea in the non-classical form. Diagnosis of CAH is made by clinical, biochemical and molecular genetic evaluation. In cases of 21-OHD, the diagnosis is based on the 17-hydroxyprogesterone (17-OHP) level being above 1000 ng/dL, measured early in the morning. In cases with borderline 17-OHP levels (200-1000 ng/dL), it is recommended to perform an adrenocorticotropic hormone (ACTH) stimulation test. Genotyping in cases with CAH should be performed if the adrenocortical profile is suspicious or if the ACTH stimulation test cannot be performed completely. After diagnosis, determining the carrier status of the parents and determining which parent the mutation was passed on from will help in interpreting the genetic results and determining the risk of recurrence in subsequent pregnancies.
21-Hydroxylase Deficiency Detected in Neonatal Screening: High Probability of False Negativity in Late Onset Form.
Despite the high sensitivity of neonatal screening in detecting the classical form of congenital adrenal hyperplasia due to 21-hydroxylase deficiency, one of the unclear issues is identifying asymptomatic children with late onset forms. The aim of this nationwide study was to analyse the association between genotype and screened level of 17-hydroxyprogesterone in patients with the late onset form of 21-hydroxylase deficiency and to quantify false negativity. In the Czech Republic, 1,866,129 neonates were screened (2006-2022). Among this cohort, 159 patients were confirmed to suffer from 21-hydroxylase deficiency, employing the 17-hydroxyprogesterone birthweight/gestational age-adjusted cut-off limits, and followed by the genetic confirmation. The screening prevalence was 1:11,737. Another 57 patients who were false negative in neonatal screening were added to this cohort based on later diagnosis by clinical suspicion. To our knowledge, such a huge nationwide cohort of false negative patients has not been documented before. Overall, 57 patients escaped from neonatal screening in the monitored period. All false negative patients had milder forms. Only one patient had simple virilising form and 56 patients had the late onset form. The probability of false negativity in the late onset form was 76.7%. The difference in 17-hydroxyprogesterone screening values was statistically significant (p<0.001) between severe forms (median 478.8 nmol/L) and milder (36.2 nmol/L) forms. Interestingly, the higher proportion of females with milder forms was statistically significant compared with the general population. A negative neonatal screening result does not exclude milder forms of 21-hydroxylase deficiency during the differential diagnostic procedure of children with precocious pseudopuberty.
📚 EuropePMCmostrando 24
Genotype-Phenotype Correlation in Children With Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency Using Next Generation Sequencing.
Molecular genetics & genomic medicineNationwide carrier screening for congenital adrenal hyperplasia: integrated approach of CYP21A2 pathogenic variant genotyping and comprehensive large gene deletion analysis.
BMC medical genomicsGenetics and Pathophysiology of Classic Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency.
The Journal of clinical endocrinology and metabolismClinical, Biochemical and Molecular Characteristics of Congenital Adrenal Hyperplasia Due to 21-hydroxylase Deficiency.
Journal of clinical research in pediatric endocrinology21-Hydroxylase Deficiency Detected in Neonatal Screening: High Probability of False Negativity in Late Onset Form.
Experimental and clinical endocrinology & diabetes : official journal, German Society of Endocrinology [and] German Diabetes AssociationGenetic Characterization of a Cohort of Italian Patients with Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency.
Molecular diagnosis & therapyAssessment of the Degree of Clinical Suspicion of 21-Hydroxylase Deficiency Prior to the Newborn Screening Result.
Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolismeGenetic and clinical characteristics including occurrence of testicular adrenal rest tumors in Slovak and Slovenian patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency.
Frontiers in endocrinologyThe underlying cause of the simple virilizing phenotype in patients with 21-hydroxylase deficiency harboring P31L variant.
Frontiers in endocrinologyGenotype-phenotype association in congenital adrenal hyperplasia due to 21-hydroxylase deficiency in children.
Clinical endocrinologyMolecular Diagnosis of Steroid 21-Hydroxylase Deficiency: A Practical Approach.
Frontiers in endocrinologyCharacteristics of In2G Variant in Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency.
Frontiers in endocrinologyFirst insights into the genetics of 21-hydroxylase deficiency in the Roma population.
Clinical endocrinology[Analysis of the degree of clinical suspect in patients with congenital adrenal hyperplasia by 21-hydroxylase deficiency before obtaining the result of the newborn screening program of the autonomous Community of Madrid.].
Revista espanola de salud publicaMolecular diagnosis of patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency.
BMC endocrine disordersSimple virilising congenital adrenal hyperplasia in monozygotic twins: A rare report and review of previous cases.
Pediatric endocrinology, diabetes, and metabolismThe Spectrum of Genetic Defects in Congenital Adrenal Hyperplasia in the Population of Cyprus: A Retrospective Analysis.
Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolismeClinical presentation and mutational spectrum in a series of 166 patients with classical 21-hydroxylase deficiency from South China.
Clinica chimica acta; international journal of clinical chemistryMortality in children with classic congenital adrenal hyperplasia and 21-hydroxylase deficiency (CAH) in Germany.
BMC endocrine disordersCongenital adrenal hyperplasia due to 21-hydroxylase deficiency in South Africa.
South African medical journal = Suid-Afrikaanse tydskrif vir geneeskundeNear-final height in 82 Chinese patients with congenital adrenal hyperplasia due to classic 21-hydroxylase deficiency: a single-center study from China.
Journal of pediatric endocrinology & metabolism : JPEMMolecular genetic analysis in 93 patients and 193 family members with classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency in Croatia.
The Journal of steroid biochemistry and molecular biology[Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: genotype-phenotype correlation].
Acta medica portuguesaSubstitution therapy in adult patients with congenital adrenal hyperplasia.
Best practice & research. Clinical endocrinology & metabolismAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Hiperplasia suprarrenal congênita clássica por deficiência de 21-hidroxilase, forma virilizante simples.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Hiperplasia suprarrenal congênita clássica por deficiência de 21-hidroxilase, forma virilizante simples
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Genotype-Phenotype Correlation in Children With Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency Using Next Generation Sequencing.
- Nationwide carrier screening for congenital adrenal hyperplasia: integrated approach of CYP21A2 pathogenic variant genotyping and comprehensive large gene deletion analysis.
- Genetics and Pathophysiology of Classic Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency.
- Clinical, Biochemical and Molecular Characteristics of Congenital Adrenal Hyperplasia Due to 21-hydroxylase Deficiency.
- 21-Hydroxylase Deficiency Detected in Neonatal Screening: High Probability of False Negativity in Late Onset Form.Experimental and clinical endocrinology & diabetes : official journal, German Society of Endocrinology [and] German Diabetes Association· 2025· PMID 39357842mais citado
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:315311(Orphanet)
- MONDO:0017840(MONDO)
- GARD:21399(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Q51797077(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Hiperplasia suprarrenal congênita clássica por deficiência de 21-hidroxilase, forma virilizante simples
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos aprovados FDA
- fonte: FDA OpenFDA
- Reposicionamento
- fonte: Drug Repurposing Hub