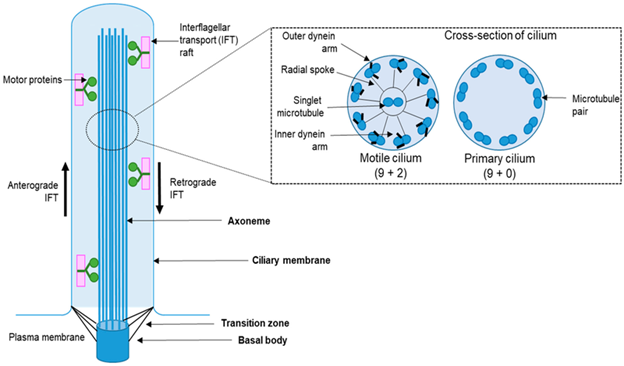
Ciliopatias são um grupo de doenças geneticamente diversas causadas por defeitos na estrutura ou na função do cílio primário, uma organela altamente especializada e evolutivamente conservada encontrada em quase todas as células eucarióticas. O cílio primário desempenha um papel central na regulação da transdução de sinal, tornando-o essencial para inúmeros processos de desenvolvimento e fisiológicos.
Introdução
O que você precisa saber de cara
Visão geral
A Nefronoftise de início tardio é uma doença renal hereditária rara que faz parte do grupo das nefronoftises. Ela se caracteriza pelo desenvolvimento de sintomas renais em uma idade mais avançada do que as formas clássicas da doença, geralmente após a infância ou na vida adulta. A condição leva a uma perda progressiva da função dos rins, podendo evoluir para insuficiência renal.[1]
Sinais e sintomas
Os sinais e sintomas da Nefronoftise de início tardio são semelhantes aos de outras formas de nefronoftise, mas aparecem mais tardiamente. Eles incluem poliúria (aumento do volume de urina), polidipsia (sede excessiva), anemia e, eventualmente, insuficiência renal. Como a doença progride lentamente, os sintomas podem ser sutis no início.[1]
Causas genéticas
A Nefronoftise de início tardio é causada por alterações (mutações) em genes específicos. Os genes associados a esta condição são: NPHP3 (que produz a proteína Nefrocistina-3), XPNPEP3 (que produz a Xaa-Pro aminopeptidase 3) e MAPKBP1 (que produz a proteína de ligação à quinase ativada por mitógeno 1). Essas mutações afetam a estrutura e a função dos cílios primários das células renais, levando à formação de cistos e à fibrose dos rins.[1][3]
Diagnóstico
O diagnóstico da Nefronoftise de início tardio é baseado na avaliação clínica, exames de imagem (como ultrassom renal) e, principalmente, em testes genéticos. O sequenciamento completo do exoma (WES) é um dos métodos disponíveis para identificar mutações nos genes associados. Além disso, exames como cariótipo (bandas G, Q ou R) e pesquisa de microdeleções/microduplicações por FISH podem ser realizados para descartar outras condições. A dosagem de alfa-fetoproteína também pode ser solicitada como parte da investigação. Atualmente, existem 336 testes genéticos disponíveis e 347 variantes registradas no ClinVar para esta doença.[1][3]
Tratamento e manejo
O tratamento da Nefronoftise de início tardio é focado no manejo dos sintomas e na desaceleração da progressão da doença renal. Não há cura específica. As medidas incluem controle da pressão arterial, tratamento da anemia e, em estágios avançados, terapia de substituição renal (diálise ou transplante renal). O acompanhamento com nefrologista é essencial. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para esta condição, incluindo atendimento em reabilitação para doenças raras.[1]
Prognóstico e qualidade de vida
O prognóstico da Nefronoftise de início tardio varia conforme a idade de início dos sintomas e a rapidez da progressão para insuficiência renal. Com o manejo adequado, incluindo diálise ou transplante renal quando necessário, muitos pacientes podem manter uma qualidade de vida razoável. O acompanhamento multidisciplinar é importante para lidar com as complicações da doença renal crônica.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Ciliopatias são um grupo de doenças geneticamente diversas causadas por defeitos na estrutura ou na função do cílio primário, uma organela altamente especializada e evolutivamente conservada encontrada em quase todas as células eucarióticas. O cílio primário desempenha um papel central na regulação da transdução de sinal, tornando-o essencial para inúmeros processos de desenvolvimento e fisiológicos.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Nefronoftise de início tardio é uma doença renal hereditária rara que faz parte do grupo das nefronoftises. Ela se caracteriza pelo desenvolvimento de sintomas renais em uma idade mais avançada do que as formas clássicas da doença, geralmente após a infância ou na vida adulta. A condição leva a uma perda progressiva da função dos rins, podendo evoluir para insuficiência renal.[1]
Sinais e sintomas
Os sinais e sintomas da Nefronoftise de início tardio são semelhantes aos de outras formas de nefronoftise, mas aparecem mais tardiamente. Eles incluem poliúria (aumento do volume de urina), polidipsia (sede excessiva), anemia e, eventualmente, insuficiência renal. Como a doença progride lentamente, os sintomas podem ser sutis no início.[1]
Causas genéticas
A Nefronoftise de início tardio é causada por alterações (mutações) em genes específicos. Os genes associados a esta condição são: NPHP3 (que produz a proteína Nefrocistina-3), XPNPEP3 (que produz a Xaa-Pro aminopeptidase 3) e MAPKBP1 (que produz a proteína de ligação à quinase ativada por mitógeno 1). Essas mutações afetam a estrutura e a função dos cílios primários das células renais, levando à formação de cistos e à fibrose dos rins.[1][3]
Diagnóstico
O diagnóstico da Nefronoftise de início tardio é baseado na avaliação clínica, exames de imagem (como ultrassom renal) e, principalmente, em testes genéticos. O sequenciamento completo do exoma (WES) é um dos métodos disponíveis para identificar mutações nos genes associados. Além disso, exames como cariótipo (bandas G, Q ou R) e pesquisa de microdeleções/microduplicações por FISH podem ser realizados para descartar outras condições. A dosagem de alfa-fetoproteína também pode ser solicitada como parte da investigação. Atualmente, existem 336 testes genéticos disponíveis e 347 variantes registradas no ClinVar para esta doença.[1][3]
Tratamento e manejo
O tratamento da Nefronoftise de início tardio é focado no manejo dos sintomas e na desaceleração da progressão da doença renal. Não há cura específica. As medidas incluem controle da pressão arterial, tratamento da anemia e, em estágios avançados, terapia de substituição renal (diálise ou transplante renal). O acompanhamento com nefrologista é essencial. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para esta condição, incluindo atendimento em reabilitação para doenças raras.[1]
Prognóstico e qualidade de vida
O prognóstico da Nefronoftise de início tardio varia conforme a idade de início dos sintomas e a rapidez da progressão para insuficiência renal. Com o manejo adequado, incluindo diálise ou transplante renal quando necessário, muitos pacientes podem manter uma qualidade de vida razoável. O acompanhamento multidisciplinar é importante para lidar com as complicações da doença renal crônica.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
3 genes identificados com associação a esta condição.
Required for normal ciliary development and function. Inhibits disheveled-1-induced canonical Wnt-signaling activity and may also play a role in the control of non-canonical Wnt signaling which regulates planar cell polarity. Probably acts as a molecular switch between different Wnt signaling pathways. Required for proper convergent extension cell movements
Cell projection, cilium
Nephronophthisis 3
An autosomal recessive disorder resulting in end-stage renal disease. It is characterized by polyuria, polydipsia, anemia. Onset of terminal renal failure occurr significantly later (median age, 19 years) than in juvenile nephronophthisis. Renal pathology is characterized by alterations of tubular basement membranes, tubular atrophy and dilation, sclerosing tubulointerstitial nephropathy, and renal cyst development predominantly at the corticomedullary junction.
Catalyzes the removal of a penultimate prolyl residue from the N-termini of peptides, such as Leu-Pro-Ala (PubMed:25609706, PubMed:28476889). Also shows low activity towards peptides with Ala or Ser at the P1 position (PubMed:28476889) Promotes TNFRSF1B-mediated phosphorylation of MAPK8/JNK1 and MAPK9/JNK2, suggesting a function as an adapter protein for TNFRSF1B; the effect is independent of XPNPEP3 peptidase activity. May inhibit apoptotic cell death induced via TNF-TNFRSF1B signaling
MitochondrionCytoplasm
Nephronophthisis-like nephropathy 1
An autosomal recessive disorder with features of nephronophthisis, a cystic kidney disease leading to end-stage renal failure. Nephronophthisis is histologically characterized by modifications of the tubules with thickening of the basement membrane, interstitial fibrosis and, in the advanced stages, medullary cysts. Typical clinical manifestation are chronic renal failure, anemia, polyuria, polydipsia, isosthenuria, and growth retardation. Associations with extrarenal symptoms are frequent. In NPHPL1 patients, extrarenal symptoms include hypertension, essential tremor, sensorineural hearing loss and gout. Severely affected individuals can manifest a mitochondrial disorder with isolated complex I deficiency activity in muscle, seizures, intellectual disability and hypertrophic dilated cardiomyopathy.
Negative regulator of NOD2 function. It down-regulates NOD2-induced processes such as activation of NF-kappa-B signaling, IL8 secretion and antibacterial response (PubMed:22700971). Involved in JNK signaling pathway (By similarity)
CytoplasmNucleusCytoplasm, cytoskeleton, spindle pole
Nephronophthisis 20
A form of nephronophthisis, an autosomal recessive chronic tubulo-interstitial nephritis that progresses to end-stage renal failure. Some patients have cystic kidneys of normal size and no extrarenal manifestations, whereas others have enlarged renal size and severe extrarenal defects, including hypertrophic obstructive cardiomyopathy, aortic stenosis, pulmonary stenosis, patent ductus arteriosus, situs inversus, and periportal liver fibrosis. NPHP20 patients do not show extrarenal manifestations or evidence of a ciliopathy, such as situs inversus or polydactyly.
Variantes genéticas (ClinVar)
347 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Nefronoftise de início tardio
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Structure-Activity Analysis Reveals Perturbed Cilia-Jun N-Terminal Kinase Signaling in MAPKBP1-Associated Kidney Disease.
Este estudo detalha a nefronoptise de início tardio (NPH) associada à perda da proteína MAPKBP1 (NPHP20), revelando uma doença renal predominantemente não-sindrômica com progressão excepcionalmente lenta, o que é crucial para o prognóstico de pacientes e o manejo clínico. A pesquisa demonstra que a disfunção de MAPKBP1 perturba os cílios primários através de uma via de sinalização JNK alterada, levando ao encurtamento ciliar. Contudo, a restauração do comprimento ciliar por meio da modulação da JNK ou de seu alvo (actina) aponta para novas abordagens terapêuticas, destacando a via JNK como um alvo promissor para futuros tratamentos da NPH.
🇧🇷 traduzidoDelayed-Onset Renal Allograft Compartment Syndrome in a Pediatric Kidney Transplant Recipient: The Role of Surgical Re-Evaluation.
Este artigo destaca que a síndrome compartimental do enxerto renal (RACS) de início tardio é uma causa subdiagnosticada de disfunção renal pós-transplante, podendo ocorrer bem após o período imediato. Para médicos e pacientes, o caso de uma adolescente ilustra que, diante de uma piora inexplicável da função do enxerto e falha de tratamentos padrão, a exploração cirúrgica é essencial. Mesmo sem anomalias visíveis no órgão, a identificação de edema tecidual circundante e a intervenção cirúrgica podem restaurar a função renal, sendo uma ferramenta diagnóstica e terapêutica vital.
🇧🇷 traduzidoThe genetic landscape and clinical spectrum of nephronophthisis and related ciliopathies.
Este estudo abrangente sobre nefronofotise (NPH), uma doença renal genética que causa insuficiência renal, revela que uma proporção considerável (34%) dos pacientes apresenta a forma de início tardio, desenvolvendo-se após os 15 anos. Essa forma tardia é frequentemente ligada a variantes genéticas específicas e pode ter menos manifestações fora dos rins do que as formas infantis, sugerindo que a NPH de início tardio está provavelmente subdiagnosticada em adultos com doença renal crônica, um ponto crucial para médicos e pacientes.
🇧🇷 traduzidoBiallelic ANKS6 mutations cause late-onset ciliopathy with chronic kidney disease through YAP dysregulation.
Este estudo identifica mutações no gene ANKS6 como uma nova causa genética de nefronoptise de início tardio, uma forma de doença renal crônica hereditária. A pesquisa revela que a deficiência da proteína ANKS6 desregula vias de sinalização cruciais (como a via Hippo e Wnt), afetando a função celular e a integridade dos cílios primários. Essas descobertas são fundamentais para entender os mecanismos da doença e abrem caminho para potenciais novos alvos terapêuticos e estratégias diagnósticas para pacientes.
🇧🇷 traduzidoWhole Exome Sequencing Reveals a XPNPEP3 Novel Mutation Causing Nephronophthisis in a Pediatric Patient.
Este estudo identificou uma nova mutação no gene XPNPEP3 como causa de Nefronoptise (NPHP), uma doença renal progressiva. É importante notar que, embora mutações em XPNPEP3 sejam geralmente associadas à NPHP de início tardio, esta mutação específica pode manifestar a doença já na primeira infância. Isso reforça a necessidade de considerar o sequenciamento genético (WES) para pacientes pediátricos com suspeita de NPHP, mesmo quando exames rotineiros não são conclusivos, a fim de obter um diagnóstico preciso e precoce, que pode ser atípico para o gene envolvido.
🇧🇷 traduzidoPublicações recentes
Effusive-constrictive pericarditis in a patient with late-onset systemic lupus erythematosus.
Thrombosed epic mitral bioprosthesis in the late postoperative period: A case report.
Clinical and molecular characteristics of early-onset pancreatic and biliary cancers.
Prevalence and risk factors for neonatal sepsis among very preterm infants in China: a systematic review and meta-analysis.
HLA genotyping and clinical characteristics of early-onset and late-onset anti-LGI1 encephalitis: a single-center cohort study in China.
📚 EuropePMCmostrando 9
Structure-Activity Analysis Reveals Perturbed Cilia-Jun N-Terminal Kinase Signaling in MAPKBP1-Associated Kidney Disease.
Kidney international reportsDelayed-Onset Renal Allograft Compartment Syndrome in a Pediatric Kidney Transplant Recipient: The Role of Surgical Re-Evaluation.
Pediatric transplantationThe genetic landscape and clinical spectrum of nephronophthisis and related ciliopathies.
Kidney internationalBiallelic ANKS6 mutations cause late-onset ciliopathy with chronic kidney disease through YAP dysregulation.
Human molecular geneticsWhole Exome Sequencing Reveals a XPNPEP3 Novel Mutation Causing Nephronophthisis in a Pediatric Patient.
Iranian biomedical journalNovel nephronophthisis-associated variants reveal functional importance of MAPKBP1 dimerization for centriolar recruitment.
Kidney internationalTwo novel homozygous mutations in NPHP1 lead to late onset end-stage renal disease: a case report of an adult nephronophthisis in a Chinese intermarriage family.
BMC nephrologyRecent advances in the molecular diagnosis of polycystic kidney disease.
Expert review of molecular diagnosticsMutations in MAPKBP1 Cause Juvenile or Late-Onset Cilia-Independent Nephronophthisis.
American journal of human geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Nefronoftise de início tardio.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Nefronoftise de início tardio
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Structure-Activity Analysis Reveals Perturbed Cilia-Jun N-Terminal Kinase Signaling in MAPKBP1-Associated Kidney Disease.
- Delayed-Onset Renal Allograft Compartment Syndrome in a Pediatric Kidney Transplant Recipient: The Role of Surgical Re-Evaluation.
- The genetic landscape and clinical spectrum of nephronophthisis and related ciliopathies.
- Biallelic ANKS6 mutations cause late-onset ciliopathy with chronic kidney disease through YAP dysregulation.
- Whole Exome Sequencing Reveals a XPNPEP3 Novel Mutation Causing Nephronophthisis in a Pediatric Patient.
- Effusive-constrictive pericarditis in a patient with late-onset systemic lupus erythematosus.
- Thrombosed epic mitral bioprosthesis in the late postoperative period: A case report.
- Clinical and molecular characteristics of early-onset pancreatic and biliary cancers.
- Prevalence and risk factors for neonatal sepsis among very preterm infants in China: a systematic review and meta-analysis.
- HLA genotyping and clinical characteristics of early-onset and late-onset anti-LGI1 encephalitis: a single-center cohort study in China.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:93589(Orphanet)
- MONDO:0019742(MONDO)
- GARD:16824(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55788850(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Nefronoftise de início tardio
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata