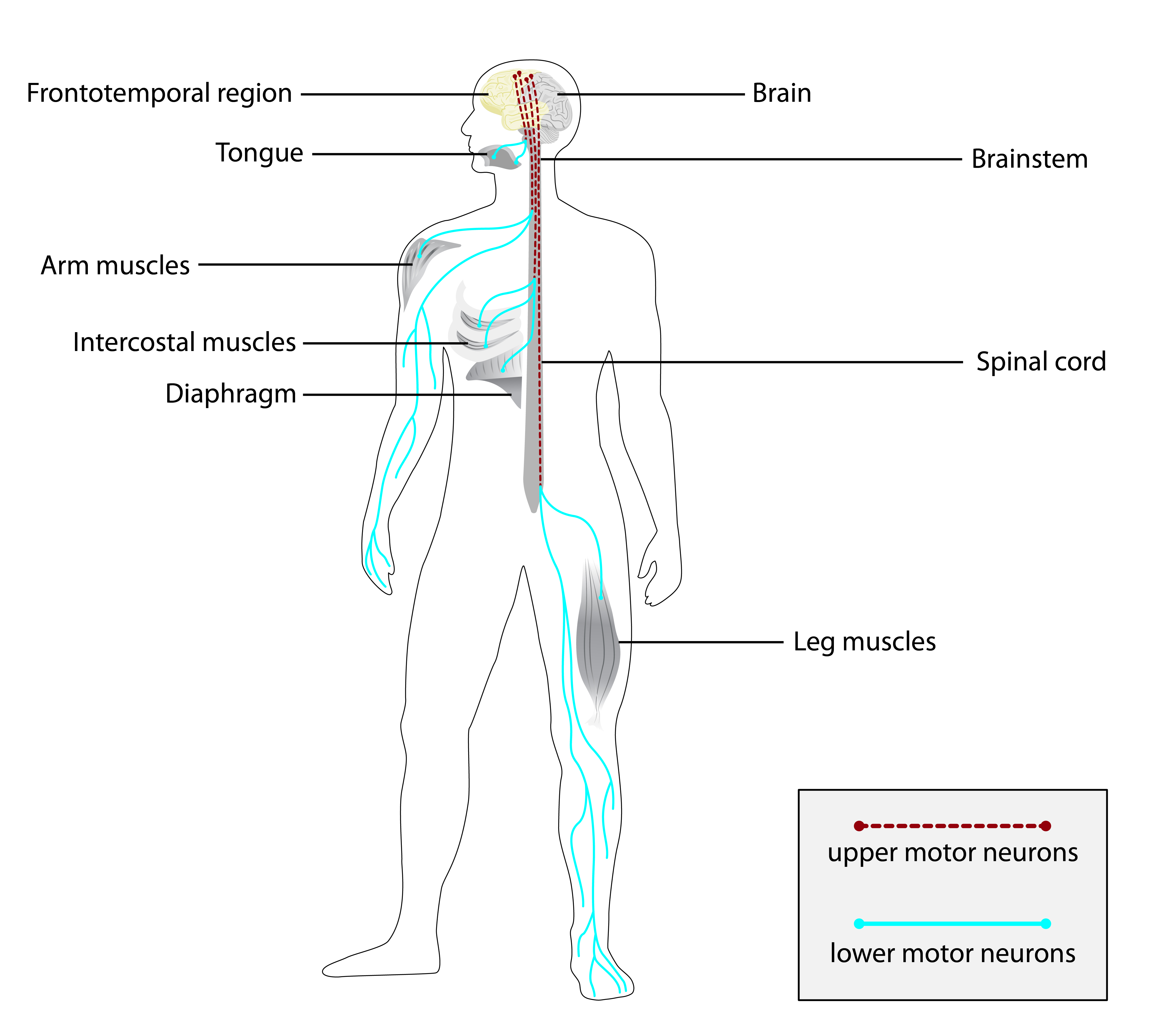
A esclerose lateral amiotrófica (ELA), também conhecida como doença do neurônio motor (DNM) ou doença de Lou Gehrig, é uma doença neurodegenerativa rara e terminal, definida pela perda progressiva dos neurônios motores superiores e inferiores que normalmente controlam a contração muscular voluntária. A ELA é a mais comum das doenças do neurônio motor. A ELA frequentemente se manifesta em seus estágios iniciais com rigidez muscular gradual, espasmos, fraqueza e atrofia. A perda de neurônios motores normalmente continua até que a capacidade de comer, falar, mover-se e respirar sem suporte mecânico seja perdida. Estima-se que pelo menos 50% das pessoas com ELA apresentem mudanças significativas no pensamento e no comportamento, sendo que 15% dos indivíduos desenvolvem demência frontotemporal.
Introdução
O que você precisa saber de cara
Visão geral
A Neuropatia axonal congênita com encefalopatia é uma doença neurológica rara, de origem genética, que se manifesta já ao nascimento ou nos primeiros dias de vida. Estima-se que afete menos de 1 em cada 1.000.000 de pessoas. A condição combina problemas nos nervos periféricos (neuropatia axonal) com comprometimento do sistema nervoso central (encefalopatia), resultando em fraqueza muscular generalizada e atraso no desenvolvimento neuropsicomotor.[1]
Sinais e sintomas
Os sintomas estão presentes desde o nascimento ou aparecem logo após. Os principais incluem: tônus muscular muito baixo (hipotonia generalizada), fraqueza muscular que é mais acentuada nas partes distais dos membros (mãos e pés), dificuldades para respirar e engolir, e ausência de reflexos tendíneos (arreflexia difusa). O envolvimento do sistema nervoso central se manifesta por microcefalia progressiva (cabeça menor que o esperado), crises epilépticas (convulsões) e atraso global do desenvolvimento (a criança não atinge os marcos esperados para a idade, como sentar, engatinhar ou falar). Outras alterações que podem estar presentes incluem: perda auditiva, lesões oculares, anomalias esqueléticas (como pé torto congênito, dedos sobrepostos, escoliose e contraturas articulares), criptorquidia (testículo não descido) e características faciais atípicas (face grosseira, hipertelorismo — olhos mais afastados — e palato em ogiva).[1]
Causas genéticas
A doença é causada por alterações (mutações) em genes ainda não especificados publicamente nas bases de dados consultadas. Sabe-se que o padrão de herança é autossômico recessivo, ou seja, é necessário herdar uma cópia do gene alterado de cada um dos pais para manifestar a doença. A base genética exata continua em investigação.[1][3]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sintomas característicos e pode ser confirmado por meio de testes genéticos. O sequenciamento completo do exoma (WES) é um dos procedimentos disponíveis no Sistema Único de Saúde (SUS) para investigação diagnóstica. Até o momento, 84 variantes genéticas associadas à doença foram registradas em bancos de dados internacionais (ClinVar).[1][3]
Tratamento e manejo
Não existe cura específica para a Neuropatia axonal congênita com encefalopatia. O tratamento é de suporte e multidisciplinar, visando aliviar os sintomas e melhorar a qualidade de vida. O manejo inclui atendimento em reabilitação para doenças raras, disponível no SUS, com fisioterapia, terapia ocupacional e fonoaudiologia. Cuidados respiratórios e nutricionais são essenciais, especialmente para lidar com as dificuldades de deglutição e o risco de pneumonia aspirativa. O controle das crises epilépticas deve ser feito com medicamentos anticonvulsivantes, sempre sob prescrição e acompanhamento médico. O suporte ortopédico pode ser necessário para contraturas e escoliose.[1]
Prognóstico e qualidade de vida
O prognóstico geralmente é reservado. A evolução da doença é frequentemente desfavorável devido à insuficiência respiratória e/ou pneumonia por aspiração, complicações comuns em crianças com fraqueza muscular grave e dificuldades de deglutição. O acompanhamento próximo por uma equipe multidisciplinar é fundamental para oferecer suporte à criança e à família, maximizando o conforto e a qualidade de vida dentro das limitações impostas pela condição.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
A esclerose lateral amiotrófica (ELA), também conhecida como doença do neurônio motor (DNM) ou doença de Lou Gehrig, é uma doença neurodegenerativa rara e terminal, definida pela perda progressiva dos neurônios motores superiores e inferiores que normalmente controlam a contração muscular voluntária. A ELA é a mais comum das doenças do neurônio motor. A ELA frequentemente se manifesta em seus estágios iniciais com rigidez muscular gradual, espasmos, fraqueza e atrofia. A perda de neurônios motores normalmente continua até que a capacidade de comer, falar, mover-se e respirar sem suporte mecânico seja perdida. Estima-se que pelo menos 50% das pessoas com ELA apresentem mudanças significativas no pensamento e no comportamento, sendo que 15% dos indivíduos desenvolvem demência frontotemporal.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Neuropatia axonal congênita com encefalopatia é uma doença neurológica rara, de origem genética, que se manifesta já ao nascimento ou nos primeiros dias de vida. Estima-se que afete menos de 1 em cada 1.000.000 de pessoas. A condição combina problemas nos nervos periféricos (neuropatia axonal) com comprometimento do sistema nervoso central (encefalopatia), resultando em fraqueza muscular generalizada e atraso no desenvolvimento neuropsicomotor.[1]
Sinais e sintomas
Os sintomas estão presentes desde o nascimento ou aparecem logo após. Os principais incluem: tônus muscular muito baixo (hipotonia generalizada), fraqueza muscular que é mais acentuada nas partes distais dos membros (mãos e pés), dificuldades para respirar e engolir, e ausência de reflexos tendíneos (arreflexia difusa). O envolvimento do sistema nervoso central se manifesta por microcefalia progressiva (cabeça menor que o esperado), crises epilépticas (convulsões) e atraso global do desenvolvimento (a criança não atinge os marcos esperados para a idade, como sentar, engatinhar ou falar). Outras alterações que podem estar presentes incluem: perda auditiva, lesões oculares, anomalias esqueléticas (como pé torto congênito, dedos sobrepostos, escoliose e contraturas articulares), criptorquidia (testículo não descido) e características faciais atípicas (face grosseira, hipertelorismo — olhos mais afastados — e palato em ogiva).[1]
Causas genéticas
A doença é causada por alterações (mutações) em genes ainda não especificados publicamente nas bases de dados consultadas. Sabe-se que o padrão de herança é autossômico recessivo, ou seja, é necessário herdar uma cópia do gene alterado de cada um dos pais para manifestar a doença. A base genética exata continua em investigação.[1][3]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sintomas característicos e pode ser confirmado por meio de testes genéticos. O sequenciamento completo do exoma (WES) é um dos procedimentos disponíveis no Sistema Único de Saúde (SUS) para investigação diagnóstica. Até o momento, 84 variantes genéticas associadas à doença foram registradas em bancos de dados internacionais (ClinVar).[1][3]
Tratamento e manejo
Não existe cura específica para a Neuropatia axonal congênita com encefalopatia. O tratamento é de suporte e multidisciplinar, visando aliviar os sintomas e melhorar a qualidade de vida. O manejo inclui atendimento em reabilitação para doenças raras, disponível no SUS, com fisioterapia, terapia ocupacional e fonoaudiologia. Cuidados respiratórios e nutricionais são essenciais, especialmente para lidar com as dificuldades de deglutição e o risco de pneumonia aspirativa. O controle das crises epilépticas deve ser feito com medicamentos anticonvulsivantes, sempre sob prescrição e acompanhamento médico. O suporte ortopédico pode ser necessário para contraturas e escoliose.[1]
Prognóstico e qualidade de vida
O prognóstico geralmente é reservado. A evolução da doença é frequentemente desfavorável devido à insuficiência respiratória e/ou pneumonia por aspiração, complicações comuns em crianças com fraqueza muscular grave e dificuldades de deglutição. O acompanhamento próximo por uma equipe multidisciplinar é fundamental para oferecer suporte à criança e à família, maximizando o conforto e a qualidade de vida dentro das limitações impostas pela condição.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Neuropatia axonal congênita com encefalopatia
Centros de Referência SUS
24 centros habilitados pelo SUS para Neuropatia axonal congênita com encefalopatia
Centros para Neuropatia axonal congênita com encefalopatia
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
ATP6AP2-Related Disease Caused by Splicing Defects: Abnormal Glycosylation and the First Affected Female.
ATP6AP2 splicing variants cause syndromic X-linked intellectual disability Hedera type (XPDS; OMIM#300423) and X-linked parkinsonism with spasticity (MRXSH; OMIM#300911). Alternatively, ATP6AP2 missense variants lead to hepatopathy, immunological abnormalities, cutis laxa and only mild intellectual disability with N-/O-glycosylation defects (ATP6AP2-CDG; OMIM#301045). The disparity between neurological and hepatic ATP6AP2-related disease entities is an ongoing puzzle. We aimed to investigate whether patients with an isolated neurological presentation of ATP6AP2-related disease, consistent with XPDS/MRXSH, also have abnormal glycosylation biomarkers, potentially implicating this as part of the pathological mechanism. We identified three males and one female from three families with ATP6AP2 splicing variants and ID/DD, epilepsy, axial hypotonia, axonal neuropathy and microcephaly; the heterozygous female has a milder phenotype. RNA-Seq in patient-derived fibroblasts validated defective splicing, correlated with lowered ATP6AP2 protein levels in fibroblasts alongside glycosylation abnormalities. We describe defective glycosylation alongside ATP6AP2 splicing variants in four patients, including the first female with ATP6AP2-related disease. This connects more closely the phenotypes of XPDS/MRXSH and ATP6AP2-CDG and indicates that abnormal glycosylation markers may be a consistent feature of splicing variants, and potentially part of the pathological mechanism underlying ATP6AP2-related disease caused by abnormal splicing. We also provide additional evidence that neurodevelopment is uniquely sensitive to the gene dosage of ATP6AP2, linked to the isolated neurological phenotype found in patients with splice variants and the attenuated, but still severe, phenotype of the female in our study. Glycosylation defects can be found in "splicing" forms of ATP6AP2-related diseases, bridging the gap between XPDS, MRXSH and ATP6AP2-CDG.
The expanding clinical and genetic spectrum of DYNC1H1-related disorders.
Intracellular trafficking involves an intricate machinery of motor complexes, including the dynein complex, to shuttle cargo for autophagolysosomal degradation. Deficiency in dynein axonemal chains, as well as cytoplasmic light and intermediate chains, have been linked with ciliary dyskinesia and skeletal dysplasia. The cytoplasmic dynein 1 heavy chain protein (DYNC1H1) serves as a core complex for retrograde trafficking in neuronal axons. Dominant pathogenic variants in DYNC1H1 have been previously implicated in peripheral neuromuscular disorders (NMD) and neurodevelopmental disorders (NDD). As heavy-chain dynein is ubiquitously expressed, the apparent selectivity of heavy chain dyneinopathy for motor neuronal phenotypes remains currently unaccounted for. Here, we aimed to evaluate the full DYNC1H1-related clinical, molecular and imaging spectrum, including multisystem features and novel phenotypes presenting throughout life. We identified 47 cases from 43 families with pathogenic heterozygous variants in DYNC1H1 (aged 0-59 years) and collected phenotypic data via a comprehensive standardized survey and clinical follow-up appointments. Most patients presented with divergent and previously unrecognized neurological and multisystem features, leading to significant delays in genetic testing and establishing the correct diagnosis. Neurological phenotypes include novel autonomic features, previously rarely described behavioral disorders, movement disorders and periventricular lesions. Sensory neuropathy was identified in nine patients (median age of onset 10.6 years), of which five were only diagnosed after the second decade of life, and three had a progressive age-dependent sensory neuropathy. Novel multisystem features included primary immunodeficiency, bilateral sensorineural hearing loss, organ anomalies and skeletal manifestations, resembling the phenotypic spectrum of other dyneinopathies. We also identified an age-dependent biphasic disease course with developmental regression in the first decade and, following a period of stability, neurodegenerative progression after the second decade of life. Of note, we observed several cases in whom neurodegeneration appeared to be prompted by intercurrent systemic infections with double-stranded DNA viruses (Herpesviridae) or single-stranded RNA viruses (Ross River fever, SARS-CoV-2). Moreover, the disease course appeared to be exacerbated by viral infections regardless of age and/or severity of neurodevelopmental disorder manifestations, indicating a role of dynein in anti-viral immunity and neuronal health. In summary, our findings expand the clinical, imaging and molecular spectrum of pathogenic DYNC1H1 variants beyond motor neuropathy disorders and suggest a life-long continuum and age-related progression due to deficient intracellular trafficking. This study will facilitate early diagnosis and improve counselling and health surveillance of affected patients.
Progressive Optic Neuropathy in Hydrocephalic Ccdc13 Mutant Mice Caused by Impaired Axoplasmic Transport at the Optic Nerve Head.
Optic nerve head (ONH) atrophy is frequently associated with hydrocephalic conditions. Cerebrospinal fluid (CSF)-containing meninges form a subarachnoid space that terminates at the ONH, which physically impacts it. This study aims to characterize optic neuropathy in congenital hydrocephalic mice with genetic disruption of the Ccdc13 gene. The ccdc13 germline knockout mice were generated. The hydrocephalus phenotype and subarachnoid space surrounding the optic nerve were evaluated using routine histology and Evans blue stain. Optic neuropathy was examined with immunohistochemistry and transmission electron microscopy (TEM). Axon transport was indicated by cholera toxin subunit B (CTB) fluorescence conjugate. Retinal function was evaluated by electroretinography (ERG), and Ccdc13 expression was revealed by a knock-in Gfp reporter. Ccdc13 mutant mice manifested hydrocephalus at birth. ONH displacement, or negative cupping, and enlarged subarachnoid space at the optic terminus occurred as early as 1 month after birth. Intraocular pressure (IOP) was normal. Optic neuropathy was first observed at the ONH, followed by a distal-to-proximal progression of optic nerve pathology indicated by alteration of axonal ultrastructure and deposition of unphosphorylated neurofilament heavy chain. Anterograde axonal transport was also hampered. Retinal ganglion cell (RGC) function was compromised as early as postnatal day 21 (P21), along with reduced neurofilament heavy chain expression. Optic neuropathy caused by disruption of Ccd13 was non-cell autonomous, stemming from hydrocephalus with presumed high intracranial pressure (ICP), which physically impacts the ONH by increasing the translaminar pressure gradient. We provided knowledge of optic neuropathy from a congenital mouse model for hydrocephalus. The hydrocephalus in mice could damage the ONH by increasing the translaminar pressure gradient and negative cupping, leading to impairment in axoplasmic transport and RGC pathology. Our findings highlight the importance of the interplay between IOP and ICP in the development of glaucoma.
Neuropathy in ARSACS is demyelinating but without typical nerve enlargement in nerve ultrasound.
To specify peripheral nerve affection in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) by correlating high-resolution nerve ultrasound and nerve conduction studies. We assessed a cohort of 11 ARSACS patients with standardized nerve conduction studies and high-resolution ultrasound of peripheral nerves and compared nerve ultrasound findings to a healthy control group matched for age, sex, size and weight. Mean age of patients was 39.0 (± 14.1) years and disease duration at assessment 30.6 (± 12.5) years. All patients presented with a spasticity, ataxia and peripheral neuropathy. Neuropathy appeared to be primarily demyelinating in 9/11 cases and was not classifiable in 2/11 cases due to not evocable potentials. Nerve ultrasound revealed a normal ultrasound pattern sum score (UPSS) in each ARSACS patient and no significant nerve enlargement compared to the control group. Peripheral neuropathy in ARSACS showed primarily demyelinating rather than axonal characteristics and presented without nerve enlargement. As demyelinating neuropathies do commonly present enlarged nerves we recommend further genetic testing of the SACS gene in patients who present with this combination of demyelinating neuropathy without nerve enlargement. ARSACS cases that initially presented only with neuropathy without spasticity or ataxia and therefore were misdiagnosed as Charcot-Marie-Tooth disease are supporting this suggestion.
Mutations in alpha-B-crystallin cause autosomal dominant axonal Charcot-Marie-Tooth disease with congenital cataracts.
Mutations in the alpha-B-crystallin (CRYAB) gene have initially been associated with myofibrillar myopathy, dilated cardiomyopathy and cataracts. For the first time, peripheral neuropathy is reported here as a novel phenotype associated with CRYAB. Whole-exome sequencing was performed in two unrelated families with genetically unsolved axonal Charcot-Marie-Tooth disease (CMT2), assessing clinical, neurophysiological and radiological features. The pathogenic CRYAB variant c.358A>G;p.Arg120Gly was segregated in all affected patients from two unrelated families. The disease presented as late onset CMT2 (onset over 40 years) with distal sensory and motor impairment and congenital cataracts. Muscle involvement was probably associated in cases showing mild axial and diaphragmatic weakness. In all cases, nerve conduction studies demonstrated the presence of an axonal sensorimotor neuropathy along with chronic neurogenic changes on needle examination. In cases with late onset autosomal dominant CMT2 and congenital cataracts, it is recommended that CRYAB is considered for genetic testing. The identification of CRYAB mutations causing CMT2 further supports a continuous spectrum of expressivity, from myopathic to neuropathic and mixed forms, of a growing number of genes involved in protein degradation and chaperone-assisted autophagy.
Publicações recentes
Report of a novel missense TDP1 variant in a Pakistani family affected with an extremely rare disorder congenital spinocerebellar ataxia with axonal neuropathy type 1 (SCAN1).
Progressive Optic Neuropathy in Hydrocephalic Ccdc13 Mutant Mice Caused by Impaired Axoplasmic Transport at the Optic Nerve Head.
Neuropathy in ARSACS is demyelinating but without typical nerve enlargement in nerve ultrasound.
Ketogenic Diet Attenuates Refractory Epilepsy of Harel-Yoon Syndrome With ATAD3A Variants: A Case Report and Review of Literature.
Novel Variants in MPV17, PRX, GJB1, and SACS Cause Charcot-Marie-Tooth and Spastic Ataxia of Charlevoix-Saguenay Type Diseases.
📚 EuropePMC1 artigos no totalmostrando 32
ATP6AP2-Related Disease Caused by Splicing Defects: Abnormal Glycosylation and the First Affected Female.
Journal of inherited metabolic diseaseReport of a novel missense TDP1 variant in a Pakistani family affected with an extremely rare disorder congenital spinocerebellar ataxia with axonal neuropathy type 1 (SCAN1).
Molecular biology reportsProgressive Optic Neuropathy in Hydrocephalic Ccdc13 Mutant Mice Caused by Impaired Axoplasmic Transport at the Optic Nerve Head.
Investigative ophthalmology & visual scienceThe expanding clinical and genetic spectrum of DYNC1H1-related disorders.
Brain : a journal of neurologyNeuropathy in ARSACS is demyelinating but without typical nerve enlargement in nerve ultrasound.
Journal of neurologyMutations in alpha-B-crystallin cause autosomal dominant axonal Charcot-Marie-Tooth disease with congenital cataracts.
European journal of neurologyCompound heterozygous variants in MAPK8IP3 were detected in severe congenital hypotonia mimicking lethal spinal muscular atrophy.
American journal of medical genetics. Part AKetogenic Diet Attenuates Refractory Epilepsy of Harel-Yoon Syndrome With ATAD3A Variants: A Case Report and Review of Literature.
Pediatric neurologyNovel Variants in MPV17, PRX, GJB1, and SACS Cause Charcot-Marie-Tooth and Spastic Ataxia of Charlevoix-Saguenay Type Diseases.
GenesAutosomal Recessive Spastic Ataxia of Charlevoix-Saguenay due to Novel Mutations in the SACS Gene.
Journal of investigative medicine high impact case reportsLoss of non-motor kinesin KIF26A causes congenital brain malformations via dysregulated neuronal migration and axonal growth as well as apoptosis.
Developmental cellIntegrative Organelle-Based Functional Proteomics: In Silico Prediction of Impaired Functional Annotations in SACS KO Cell Model.
BiomoleculesClinical and Molecular Findings of Autosomal Recessive Spastic Ataxia of Charlevoix Saguenay: an Iranian Case Series Expanding the Genetic and Neuroimaging Spectra.
Cerebellum (London, England)Unique Ataxia-Oculomotor Apraxia 2 (AOA2) in Israel with Novel Variants, Atypical Late Presentation, and Possible Identification of a Poison Exon.
Journal of molecular neuroscience : MNPathogenic NR2F1 variants cause a developmental ocular phenotype recapitulated in a mutant mouse model.
Brain communicationsCase 293: Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay.
RadiologyComplex Movement Disorders in Ataxia with Oculomotor Apraxia Type 1: Beyond the Cerebellar Syndrome.
Tremor and other hyperkinetic movements (New York, N.Y.)KIF1A-related disorders in children: A wide spectrum of central and peripheral nervous system involvement.
Journal of the peripheral nervous system : JPNSVariants in NGLY1 lead to intellectual disability, myoclonus epilepsy, sensorimotor axonal polyneuropathy and mitochondrial dysfunction.
Clinical geneticsAn update on clinical, pathological, diagnostic, and therapeutic perspectives of childhood leukodystrophies.
Expert review of neurotherapeuticsBilateral Striatal Necrosis with Polyneuropathy with a Novel SLC25A19 (Mitochondrial Thiamine Pyrophosphate Carrier OMIMI*606521) Mutation: Treatable Thiamine Metabolic Disorder-A Report of Two Indian Cases.
NeuropediatricsExpanding the clinical description of autosomal recessive spastic ataxia of Charlevoix-Saguenay.
Journal of the neurological sciencesDisruption of Spermatogenesis and Infertility in Ataxia with Oculomotor Apraxia Type 2 (AOA2).
Cerebellum (London, England)Absence of Axoglial Paranodal Junctions in a Child With CNTNAP1 Mutations, Hypomyelination, and Arthrogryposis.
Journal of child neurologyNovel homozygous GBA2 mutation in a patient with complicated spastic paraplegia.
Clinical neurology and neurosurgeryNew spectrum of the neurologic consequences of Zika.
Journal of the neurological sciencesDemyelination, strokes, and eculizumab: Lessons from the congenital CD59 gene mutations.
Molecular immunologyXRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia.
NatureThe second report of a new hypomyelinating disease due to a defect in the VPS11 gene discloses a massive lysosomal involvement.
Journal of inherited metabolic diseaseAbnormal Glycosylation Profile and High Alpha-Fetoprotein in a Patient with Twinkle Variants.
JIMD reportsMutations in the MORC2 gene cause axonal Charcot-Marie-Tooth disease.
Brain : a journal of neurologyComplete APTX deletion in a patient with ataxia with oculomotor apraxia type 1.
BMC medical geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Neuropatia axonal congênita com encefalopatia.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Neuropatia axonal congênita com encefalopatia
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- ATP6AP2-Related Disease Caused by Splicing Defects: Abnormal Glycosylation and the First Affected Female.
- The expanding clinical and genetic spectrum of DYNC1H1-related disorders.
- Progressive Optic Neuropathy in Hydrocephalic Ccdc13 Mutant Mice Caused by Impaired Axoplasmic Transport at the Optic Nerve Head.
- Neuropathy in ARSACS is demyelinating but without typical nerve enlargement in nerve ultrasound.
- Mutations in alpha-B-crystallin cause autosomal dominant axonal Charcot-Marie-Tooth disease with congenital cataracts.
- Report of a novel missense TDP1 variant in a Pakistani family affected with an extremely rare disorder congenital spinocerebellar ataxia with axonal neuropathy type 1 (SCAN1).
- Ketogenic Diet Attenuates Refractory Epilepsy of Harel-Yoon Syndrome With ATAD3A Variants: A Case Report and Review of Literature.
- Novel Variants in MPV17, PRX, GJB1, and SACS Cause Charcot-Marie-Tooth and Spastic Ataxia of Charlevoix-Saguenay Type Diseases.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:538101(Orphanet)
- MONDO:0034041(MONDO)
- GARD:22218(GARD (NIH))
- Busca completa no PubMed(PubMed)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Neuropatia axonal congênita com encefalopatia
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)