Síndrome congênita associada à exposição pré-natal à carbamazepina, caracterizada por anomalias craniofaciais, defeitos cardíacos, atraso no desenvolvimento e outras malformações.
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome da carbamazepina fetal é uma condição que ocorre quando o embrião ou feto é exposto à carbamazepina durante a gestação. Trata-se de uma embriofetopatia relacionada a medicamentos, caracterizada por alterações na face (dismorfismo facial), com algumas semelhanças àquelas observadas na síndrome fetal do valproato. A exposição à carbamazepina, especialmente quando combinada com valproato, tem sido associada a atraso significativo no desenvolvimento (particularmente afetando a inteligência verbal) e a uma alta taxa de anomalias congênitas.[1]
Sinais e sintomas
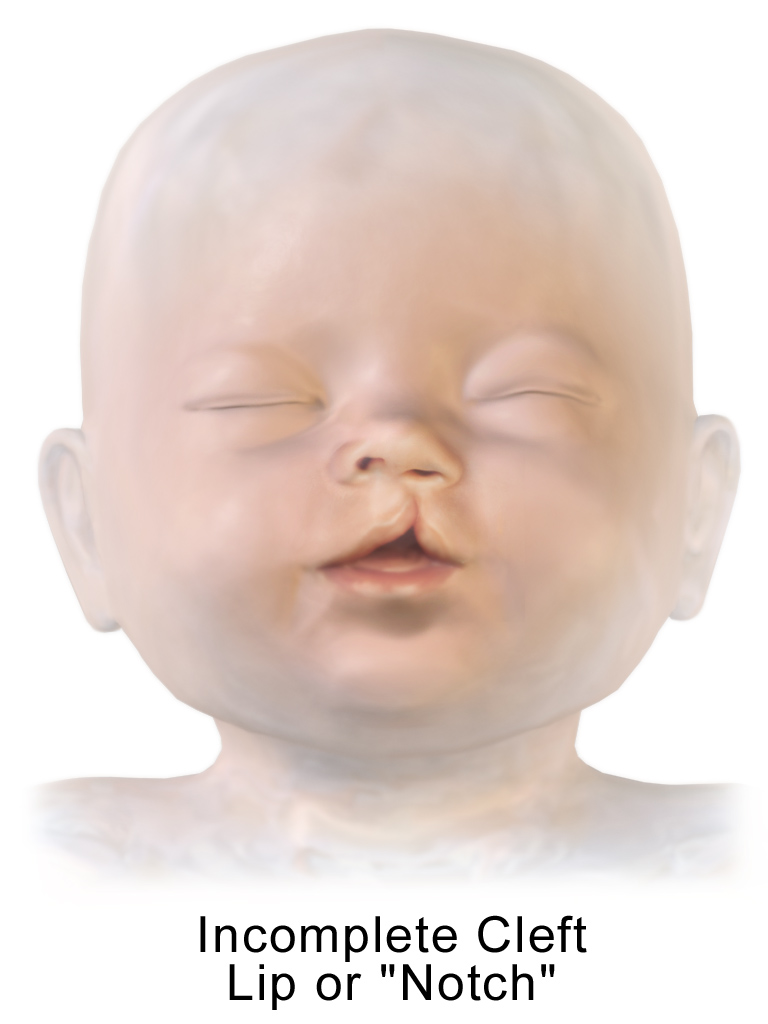
Os principais sinais incluem dismorfismo facial, como pregas epicânticas (dobra de pele no canto interno do olho), fendas palpebrais oblíquas para cima, nariz curto, micrognatia (mandíbula pequena) e hipoplasia malar (pouco desenvolvimento das maçãs do rosto). Também podem ocorrer displasia ungueal (alterações nas unhas) e anomalias maiores, como lábio leporino/fenda palatina, defeitos do tubo neural e anomalias cardíacas.[1]
Causas genéticas
Diagnóstico
O diagnóstico é baseado na história de exposição à carbamazepina durante a gestação e na presença dos sinais clínicos característicos. Exames complementares podem incluir cariótipo (bandas G, Q ou R), pesquisa de microdeleções/microduplicações por FISH, sequenciamento completo do exoma (WES) e dosagem de alfa-fetoproteína. Esses exames ajudam a descartar outras condições e a avaliar possíveis anomalias associadas.[1][3]
Tratamento e manejo
O manejo é multidisciplinar e inclui atendimento em reabilitação para doenças raras. O tratamento é direcionado aos sintomas específicos de cada paciente, podendo envolver cirurgias corretivas para anomalias como lábio leporino, fenda palatina e defeitos cardíacos, além de acompanhamento do desenvolvimento neuropsicomotor. Não há medicamentos específicos aprovados para tratar a síndrome em si.[1]
Tratamentos citados na literatura
Não há fármacos listados na literatura científica como tratamento específico para a Síndrome da carbamazepina fetal. As intervenções descritas são de suporte e correção cirúrgica das anomalias congênitas.[1]
Prognóstico e qualidade de vida
O prognóstico depende da gravidade das anomalias congênitas e do grau de atraso no desenvolvimento. A exposição combinada com valproato está associada a maior risco de comprometimento cognitivo, especialmente na área verbal. O acompanhamento precoce com equipe multidisciplinar é fundamental para otimizar o desenvolvimento e a qualidade de vida.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Síndrome congênita associada à exposição pré-natal à carbamazepina, caracterizada por anomalias craniofaciais, defeitos cardíacos, atraso no desenvolvimento e outras malformações.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome da carbamazepina fetal é uma condição que ocorre quando o embrião ou feto é exposto à carbamazepina durante a gestação. Trata-se de uma embriofetopatia relacionada a medicamentos, caracterizada por alterações na face (dismorfismo facial), com algumas semelhanças àquelas observadas na síndrome fetal do valproato. A exposição à carbamazepina, especialmente quando combinada com valproato, tem sido associada a atraso significativo no desenvolvimento (particularmente afetando a inteligência verbal) e a uma alta taxa de anomalias congênitas.[1]
Sinais e sintomas
Os principais sinais incluem dismorfismo facial, como pregas epicânticas (dobra de pele no canto interno do olho), fendas palpebrais oblíquas para cima, nariz curto, micrognatia (mandíbula pequena) e hipoplasia malar (pouco desenvolvimento das maçãs do rosto). Também podem ocorrer displasia ungueal (alterações nas unhas) e anomalias maiores, como lábio leporino/fenda palatina, defeitos do tubo neural e anomalias cardíacas.[1]
Causas genéticas
Diagnóstico
O diagnóstico é baseado na história de exposição à carbamazepina durante a gestação e na presença dos sinais clínicos característicos. Exames complementares podem incluir cariótipo (bandas G, Q ou R), pesquisa de microdeleções/microduplicações por FISH, sequenciamento completo do exoma (WES) e dosagem de alfa-fetoproteína. Esses exames ajudam a descartar outras condições e a avaliar possíveis anomalias associadas.[1][3]
Tratamento e manejo
O manejo é multidisciplinar e inclui atendimento em reabilitação para doenças raras. O tratamento é direcionado aos sintomas específicos de cada paciente, podendo envolver cirurgias corretivas para anomalias como lábio leporino, fenda palatina e defeitos cardíacos, além de acompanhamento do desenvolvimento neuropsicomotor. Não há medicamentos específicos aprovados para tratar a síndrome em si.[1]
Tratamentos citados na literatura
Não há fármacos listados na literatura científica como tratamento específico para a Síndrome da carbamazepina fetal. As intervenções descritas são de suporte e correção cirúrgica das anomalias congênitas.[1]
Prognóstico e qualidade de vida
O prognóstico depende da gravidade das anomalias congênitas e do grau de atraso no desenvolvimento. A exposição combinada com valproato está associada a maior risco de comprometimento cognitivo, especialmente na área verbal. O acompanhamento precoce com equipe multidisciplinar é fundamental para otimizar o desenvolvimento e a qualidade de vida.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome da carbamazepina fetal
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Publicações recentes
Ver todas no PubMed📚 EuropePMC1 artigos no totalmostrando 0
Ver todos os 1 no EuropePMCAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome da carbamazepina fetal.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome da carbamazepina fetal
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:370076(Orphanet)
- MONDO:0018263(MONDO)
- GARD:21595(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q55787832(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome da carbamazepina fetal
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata