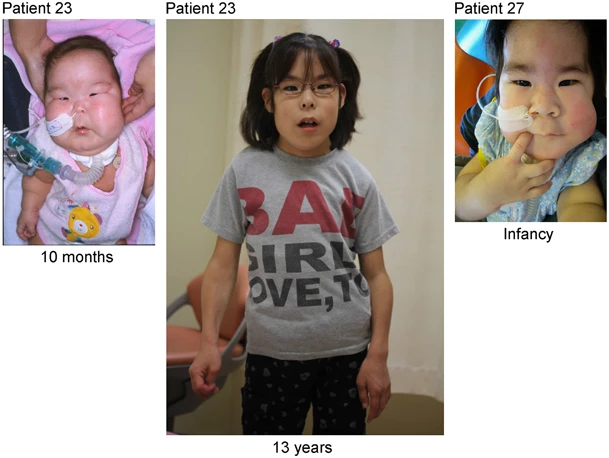
A síndrome de Kagami-Ogata é uma doença genética rara causada por mutações no cromossomo 14 materno ou por UPD(14) paterna. Os principais sinais desta doença são: polidrâmnio, tórax estreito em forma de sino, costelas em forma de cabide, defeito na parede abdominal e placenta aumentada. Pacientes com KOS também apresentam dismorfismo facial, como: bossa frontal, crescimento excessivo de pelos na testa, ponte nasal deprimida, micrognatia com ou sem retrognatia, bochechas cheias, pescoço alado e filtro labial proeminente.
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome de onfalocele familiar com dismorfia facial é uma condição genética rara, caracterizada pela presença de onfalocele (abertura na parede abdominal que permite a saída de órgãos internos) associada a características faciais distintas. A doença é considerada um defeito do desenvolvimento durante a embriogênese. Sua prevalência é estimada em menos de 1 caso por 1.000.000 de pessoas.[1]
Sinais e sintomas
Os principais sinais e sintomas incluem onfalocele, que é uma protrusão de órgãos abdominais através do umbigo, e dismorfia facial. As características faciais típicas são: face achatada, nariz curto e virado para cima, filtro nasal (sulco entre o nariz e o lábio superior) longo e largo, e arco maxilar achatado. Anormalidades nas mãos também podem estar presentes. Os sintomas geralmente se manifestam no período neonatal ou na primeira infância.[1]
Causas genéticas
A síndrome tem herança autossômica dominante, o que significa que uma cópia alterada do gene responsável (em cada célula) é suficiente para causar a condição. Até o momento, o gene específico associado a esta síndrome não foi identificado (símbolo do gene: nulo). A base genética exata permanece em estudo.[1][3]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sinais característicos (onfalocele e dismorfia facial) e pode ser complementado por exames genéticos. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para os seguintes procedimentos diagnósticos: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; sequenciamento completo do exoma (WES); e dosagem de alfa-fetoproteína. Atualmente, existem 6 testes genéticos disponíveis para a condição, mas nenhuma variante específica foi depositada no ClinVar.[1][3]
Tratamento e manejo
O manejo da síndrome é multidisciplinar e focado nos sintomas apresentados. A onfalocele geralmente requer correção cirúrgica após o nascimento. O acompanhamento com reabilitação (incluindo atendimento em reabilitação para doenças raras) é indicado para abordar possíveis atrasos no desenvolvimento ou outras necessidades funcionais. Não há medicamentos específicos aprovados para a síndrome. O tratamento deve ser individualizado, com base na avaliação de uma equipe médica especializada.[1]
Tratamentos citados na literatura
Não há fármacos ou intervenções medicamentosas listadas na literatura científica (PubTator3) especificamente para esta síndrome. Portanto, nenhum tratamento medicamentoso pode ser citado como associado à condição.[1]
Prognóstico e qualidade de vida
O prognóstico depende da gravidade da onfalocele e da presença de outras anomalias associadas. Com correção cirúrgica adequada e acompanhamento multidisciplinar, muitas crianças podem ter uma qualidade de vida satisfatória. No entanto, por ser uma condição rara e com poucos casos descritos, o prognóstico a longo prazo deve ser discutido caso a caso com o médico geneticista e a equipe de cuidados.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
A síndrome de Kagami-Ogata é uma doença genética rara causada por mutações no cromossomo 14 materno ou por UPD(14) paterna. Os principais sinais desta doença são: polidrâmnio, tórax estreito em forma de sino, costelas em forma de cabide, defeito na parede abdominal e placenta aumentada. Pacientes com KOS também apresentam dismorfismo facial, como: bossa frontal, crescimento excessivo de pelos na testa, ponte nasal deprimida, micrognatia com ou sem retrognatia, bochechas cheias, pescoço alado e filtro labial proeminente.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome de onfalocele familiar com dismorfia facial é uma condição genética rara, caracterizada pela presença de onfalocele (abertura na parede abdominal que permite a saída de órgãos internos) associada a características faciais distintas. A doença é considerada um defeito do desenvolvimento durante a embriogênese. Sua prevalência é estimada em menos de 1 caso por 1.000.000 de pessoas.[1]
Sinais e sintomas
Os principais sinais e sintomas incluem onfalocele, que é uma protrusão de órgãos abdominais através do umbigo, e dismorfia facial. As características faciais típicas são: face achatada, nariz curto e virado para cima, filtro nasal (sulco entre o nariz e o lábio superior) longo e largo, e arco maxilar achatado. Anormalidades nas mãos também podem estar presentes. Os sintomas geralmente se manifestam no período neonatal ou na primeira infância.[1]
Causas genéticas
A síndrome tem herança autossômica dominante, o que significa que uma cópia alterada do gene responsável (em cada célula) é suficiente para causar a condição. Até o momento, o gene específico associado a esta síndrome não foi identificado (símbolo do gene: nulo). A base genética exata permanece em estudo.[1][3]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sinais característicos (onfalocele e dismorfia facial) e pode ser complementado por exames genéticos. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para os seguintes procedimentos diagnósticos: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; sequenciamento completo do exoma (WES); e dosagem de alfa-fetoproteína. Atualmente, existem 6 testes genéticos disponíveis para a condição, mas nenhuma variante específica foi depositada no ClinVar.[1][3]
Tratamento e manejo
O manejo da síndrome é multidisciplinar e focado nos sintomas apresentados. A onfalocele geralmente requer correção cirúrgica após o nascimento. O acompanhamento com reabilitação (incluindo atendimento em reabilitação para doenças raras) é indicado para abordar possíveis atrasos no desenvolvimento ou outras necessidades funcionais. Não há medicamentos específicos aprovados para a síndrome. O tratamento deve ser individualizado, com base na avaliação de uma equipe médica especializada.[1]
Tratamentos citados na literatura
Não há fármacos ou intervenções medicamentosas listadas na literatura científica (PubTator3) especificamente para esta síndrome. Portanto, nenhum tratamento medicamentoso pode ser citado como associado à condição.[1]
Prognóstico e qualidade de vida
O prognóstico depende da gravidade da onfalocele e da presença de outras anomalias associadas. Com correção cirúrgica adequada e acompanhamento multidisciplinar, muitas crianças podem ter uma qualidade de vida satisfatória. No entanto, por ser uma condição rara e com poucos casos descritos, o prognóstico a longo prazo deve ser discutido caso a caso com o médico geneticista e a equipe de cuidados.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de onfalocele familiar com dismorfia facial
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Phenotype Analysis in Two Families With Otopalatodigital Syndrome Spectrum Disorder Based on FLNA Gene Variants.
Otopalatodigital spectrum disorders (OPDSD), comprising otopalatodigital syndromes types 1 and 2 (OPD1, OPD2) and frontometaphyseal dysplasia (FMD), are rare X-linked disorders caused by FLNA gene variants, with phenotypes ranging from mild skeletal anomalies to severe multisystem malformations. We describe two unrelated cases: a 14-year-old male (P1, FMD) and an aborted fetus (P2, OPD2). Whole-exome sequencing identified hemizygous maternally inherited FLNA gene variants in P1 (c.733G>A; p.Glu245Lys) and P2 (c.3707G>A; p.Gly1236Asp, novel), expanding the OPD2 mutational spectrum (NM_001110556). P1 presented with facial dysmorphism, dental anomalies, flattened thumbs, and dermatoglyphic changes; P2 showed facial dysmorphism, skeletal and cardiac malformations, and omphalocele. These cases underscore the breadth of OPDSD phenotypic variability and add novel genetic data. Dental management demands multidisciplinary care from infancy through adolescence, including cleft repair, orthodontics for micrognathia and facial aesthetics, and treatment of dental anomalies. Early recognition, molecular diagnosis, and coordinated management are critical for improving outcomes in these complex disorders.
Concomitant omphalocele, craniorachischisis and ectopic cordis associated with trisomy 18 diagnosed in first trimester.
We present a rare case of trisomy 18 of maternal origin in a pregnancy with omphalocele, craniorachischisis, and ectopia cordis. A 38-year-old woman, G3P1A1, was diagnosed with fetal anencephaly, an extrathoracic heart (ectopic cordis), deformity of spine and a stomach-and-intestine-containing omphalocele by prenatal ultrasound at 12 weeks of gestation. The patient's husband was 39 years old and healthy. The patient had no significant past medical history. She was a nonsmoker, with a pre-pregnancy BMI of 23.7 and was not diabetic. There was no family history of malformations, diseases, or teratogenic medication. The patient had not received assisted reproductive technology during this pregnancy. The pregnancy was terminated in the 13th gestational week. Additional anomalies detected after termination of the pregnancy included small and triangular face, abnormal posturing of hands, clubfoot and craniorachischisis. Crown-heel length was 5.5 cm consistent with a 12-13-week gestational age. Postnatal cytogenetic analysis of chorionic villi obtained by placental sampling revealed a karyotype of 47, XX + 18. Polymorphic DNA marker analysis by quantitative fluorescent polymerase chain reaction (QF-PCR) assays showed a maternal origin of the extra chromosome 18. The concomitant omphalocele, ectopia cordis and craniorachischisis may be related to trisomy 18. Genetic analysis of the postmortem tissue and the analysis of the parental origin of the extra chromosome 18 using QF-PCR, provide valuable information for genetic counseling. Beckwith-Wiedemann syndrome (BWS) is a growth disorder variably characterized by macroglossia, hemihyperplasia, omphalocele, neonatal hypoglycemia, macrosomia, embryonal tumors (e.g., Wilms tumor, hepatoblastoma, neuroblastoma, and rhabdomyosarcoma), visceromegaly, adrenocortical cytomegaly, kidney abnormalities (e.g., medullary dysplasia, nephrocalcinosis, and medullary sponge kidney), and ear creases / posterior helical ear pits. BWS is considered a clinical spectrum, in which affected individuals may have many or only one or two of the characteristic clinical features. Although most individuals with BWS show rapid growth in late fetal development and early childhood, growth rate usually slows by age seven to eight years. Adult heights are typically within the normal range. Hemihyperplasia (also known as lateralized overgrowth) is often appreciated at birth and may become more or less evident over time. Hemihyperplasia may affect segmental regions of the body or selected organs and tissues. Hemihyperplasia may be limited to one side of the body (ipsilateral) or involve opposite sides of the body (contralateral). Macroglossia is generally present at birth and can obstruct breathing or interfere with feeding in infants. Neonatal hypoglycemia occurs in approximately 50% of infants with BWS; most episodes are mild and transient. However, in some cases, persistent hypoglycemia due to hyperinsulinism may require consultation with an endocrinologist for therapeutic intervention. With respect to the increased risk for embryonal tumor development, the risk for Wilms tumor appears to be concentrated in the first seven years of life, whereas the risk for developing hepatoblastoma is concentrated in the first three to four years of life. Cognitive and neurobehavioral development is usually normal. After childhood, prognosis is generally favorable, although some adults experience issues requiring medical management (e.g., for renal or skeletal concerns). The clinical diagnosis of BWS can be established in a proband who has two tier 1 characteristic clinical findings OR one tier 1 and one tier 2 clinical finding. A diagnosis can also be established in a proband with at least one tier 1 or tier 2 clinical finding AND either: A constitutional epigenetic or genomic alteration leading to an abnormal methylation pattern at 11p15.5 known to be associated with BWS; OR. A copy number variant of chromosome 11p15.5 known to be associated with BWS; OR. A heterozygous BWS-causing pathogenic variant in CDKN1C. Treatment of manifestations: Hypoglycemia is treated with oral feeding if it is mild or with glucose supplementation. Hyperinsulinism is treated with standard pharmacotherapy per endocrinologist; partial pancreatectomy may be considered in those with persistent hypoglycemia who are unresponsive to pharmacologic treatment. Feeding and/or respiratory support (sometimes requiring intubation at birth or tracheostomy for severe respiratory insufficiency) may be needed for those with macroglossia and/or upper airway obstruction. Tongue reduction surgery may be considered for this and other indications. Standard treatment is recommended for omphalocele per pediatric surgeon. A shoe lift may be considered in those with a leg length discrepancy. Epiphysiodesis prior to epiphyseal closure in early puberty may be considered in instances with leg length discrepancy >2 cm; alternatively, leg lengthening of the shorter leg may be considered. For those with hemihyperplasia of the face, referral to a craniofacial center for assessment and potential treatments may be considered. If present, standard treatment is recommended for speech delay/impediment, neoplasia, congenital heart defects, and hypercalciuria / kidney anomalies. Surveillance: Tumor surveillance. Perspectives on screening for malignant tumors in childhood differ based on local, national, and international practices. In North America, proactive tumor screening is recommended when the risk of tumor development exceeds 1%. In many European countries, proactive tumor screening protocols are typically undertaken when the risk of tumor development exceeds 5% and are based on molecular mechanism. For most BWS molecular subtypes, tumor screening consists of abdominal ultrasound with views of the liver, adrenal glands, and kidneys every three months until age four years followed by kidney ultrasound only every three months from age four to seven years. Serum alpha-fetoprotein (AFP) levels are performed every three months until age four years. Physical exam by a pediatrician, geneticist, or pediatric oncologist twice a year is also recommended. Proposed screening for neuroblastoma in children with a heterozygous pathogenic variant in CDKN1C includes abdominal ultrasound, urine vanillylmandelic acid and homovanillic acid, and chest radiograph every three months until age six years, then every six months until age ten years. Non-tumor surveillance includes measurement of growth parameters, assessment for signs/symptoms of sleep apnea, and monitoring of developmental progress / educational needs at each visit; pre-feed serum glucose level per endocrinologist recommendations in neonates and infants with a history of hypoglycemia/hyperinsulinism or random serum glucose level in neonates and infants with signs/symptoms consistent with hypoglycemia; consideration of blood pressure measurements and measurement of urinary calcium-to-creatinine ratio to screen for occult nephrocalcinosis annually or biannually; consideration of kidney ultrasound to identify findings such as nephrocalcinosis and medullary sponge kidney annually between age eight years and mid-adolescence and periodically in adulthood; assessment of hemihyperplasia and leg length discrepancy at each visit at least until skeletal maturity; dental evaluation with low threshold for orthodontic evaluation as clinically indicated. BWS is associated with abnormal expression of imprinted genes in the BWS critical region. Reliable recurrence risk assessment requires identification of the genetic mechanism in the proband that underlies the abnormal expression of imprinted genes in the BWS critical region. While the majority of families have a recurrence risk of less than 1%, certain underlying genetic mechanisms (e.g., CDKN1C pathogenic variants and copy number variants involving 11p15) may be associated with a recurrence risk as high as 50% depending on the sex of the transmitting parent and the specific alteration. In families with recurrence of BWS, maternally inherited CDKN1C pathogenic variants account for approximately 40% of genetic alterations and paternally or maternally inherited copy number variants account for approximately 9% of genetic alterations. Of note, some individuals with BWS have methylation alterations in the 11p15 imprinted domain as well as in other imprinted loci. For these individuals review of the maternal history should be undertaken for unexplained spontaneous abortion, hydatidiform moles, or a sib with BWS or another imprinting disorder (e.g., Silver-Russell syndrome); in such cases, a homozygous or heterozygous pathogenic variant in maternal effect gene in the mother's genome may confer a significant recurrence risk. Prenatal testing and preimplantation genetic testing: If a genomic variant involving chromosome 11p15.5 (i.e., a cytogenetically visible duplication, inversion, or translocation), copy number variant of 11p15.5, or a CDKN1C pathogenic variant has been identified in the proband, prenatal testing via analysis of fetal DNA from samples obtained by chorionic villus sampling (CVS) or amniocentesis is possible. Preimplantation genetic testing is possible for a familial CDKN1C pathogenic variant and may be possible for some familial genomic variants. For evaluation of fetal methylation status, DNA extracted from amniotic fluid is currently felt to provide the most reliable tissue source, although false negative findings have been reported. Tissue obtained via CVS for prenatal testing for methylation status does not yield reliable results. Genetic counseling should emphasize the potential limitations of prenatal testing for epigenetic alterations.
A Male Case of Kagami-Ogata Syndrome Caused by Paternal Unipaternal Disomy 14 as a Result of a Robertsonian Translocation.
Kagami-Ogata syndrome (KOS) is a rare imprinting disorder characterized by skeletal abnormalities, dysmorphic facial features, growth retardation and developmental delay. The genetic etiology of KOS includes paternal uniparental disomy 14 [upd(14)pat], epimutations and microdeletions affecting the maternally derived imprinted region of chromosome 14q32.2. More than seventy KOS cases have been reported thus far; however, only 10, including two familial, are associated with upd(14)pat harboring Robertsonian translocation (ROB). Here, we reported a male infant with clinical manifestations of facial dysmorphism, bell-shaped small thorax, and omphalocele. Karyotype analyses identify a balanced ROB involving the long arms of chromosomes 13 and 14 both in the patient and his father. We further confirm the pattern of upd(14)pat utilizing DNA polymorphic markers. In conclusion, our case report provides a new male KOS case caused by upd(14)pat with paternally inherited Robertsonian translocation, which represents the second male case officially reported. Notably, a KOS case due to upd(14)pat and ROB is rare. An accurate diagnosis requires not only the identification of the characteristic clinical features but also systemic cytogenetic and molecular studies.
Cardiac Surgery in Trisomy 13 and 18: A Guide to Clinical Decision-Making.
There has been substantial controversy regarding treatment of congenital heart defects in infants with trisomies 13 and 18. Most reports have focused on surgical outcomes versus expectant treatment, and rarely there has been an effort to consolidate existing evidence into a more coherent way to help clinicians with decision-making and counseling families. An extensive review of the existing literature on cardiac surgery in patients with these trisomies was conducted from 2004 to 2020. The effects of preoperative and perioperative factors on in-hospital and long-term mortality were analyzed, as well as possible predictors for postoperative chronic care needs such as tracheostomy and gastrostomy. Patients with minimal or no preoperative pulmonary hypertension and mechanical ventilation undergoing corrective surgery at a weight greater than 2.5 kg suffer from lower postoperative mortality. Infants with lower-complexity cardiac defects are likely to benefit the most from surgery, although their expected mortality is higher than that of infants without trisomy. Omphalocele confers an increased mortality risk regardless of cardiac surgery. Gastrointestinal comorbidities increased the risk of gastrostomy tube placement, while those with prolonged mechanical ventilation and respiratory comorbidities are more likely to require tracheostomy. Cardiac surgery is feasible in children with trisomies 13 and 18 and can provide improved long-term results. However, this is a clinically complex population, and both physicians and caretakers should be aware of the long-term challenges these patients face following surgery when discussing treatment options.
PPP2R3C gene variants cause syndromic 46,XY gonadal dysgenesis and impaired spermatogenesis in humans.
Context Most of the knowledge on the factors involved in human sexual development stems from studies of rare cases with disorders of sex development. Here, we have described a novel 46, XY complete gonadal dysgenesis syndrome caused by homozygous variants in PPP2R3C gene. This gene encodes B″gamma regulatory subunit of the protein phosphatase 2A (PP2A), which is a serine/threonine phosphatase involved in the phospho-regulation processes of most mammalian cell types. PPP2R3C gene is most abundantly expressed in testis in humans, while its function was hitherto unknown. Patients and methods Four girls from four unrelated families with 46, XY complete gonadal dysgenesis were studied using exome or Sanger sequencing of PPP2R3C gene. In total, four patients and their heterozygous parents were investigated for clinical, laboratory, immunohistochemical and molecular characteristics. Results We have identified three different homozygous PPP2R3C variants, c.308T>C (p.L103P), c.578T>C (p.L193S) and c.1049T>C (p.F350S), in four girls with 46, XY complete gonadal dysgenesis. Patients also manifested a unique syndrome of extragonadal anomalies, including typical facial gestalt, low birth weight, myopathy, rod and cone dystrophy, anal atresia, omphalocele, sensorineural hearing loss, dry and scaly skin, skeletal abnormalities, renal agenesis and neuromotor delay. We have shown a decreased SOX9-Phospho protein expression in the dysgenetic gonads of the patients with homozygous PPP2R3C variants suggesting impaired SOX9 signaling in the pathogenesis of gonadal dysgenesis. Heterozygous males presented with abnormal sperm morphology and impaired fertility. Conclusion Our findings suggest that PPP2R3C protein is involved in the ontogeny of multiple organs, especially critical for testis development and spermatogenesis. PPPR3C provides insight into pathophysiology, as well as emerging as a potential therapeutic target for male infertility.
Publicações recentes
Phenotype Analysis in Two Families With Otopalatodigital Syndrome Spectrum Disorder Based on FLNA Gene Variants.
Concomitant omphalocele, craniorachischisis and ectopic cordis associated with trisomy 18 diagnosed in first trimester.
Cardiac Surgery in Trisomy 13 and 18: A Guide to Clinical Decision-Making.
PPP2R3C gene variants cause syndromic 46,XY gonadal dysgenesis and impaired spermatogenesis in humans.
Phenotypic spectrum associated with SPECC1L pathogenic variants: new families and critical review of the nosology of Teebi, Opitz GBBB, and Baraitser-Winter syndromes.
📚 EuropePMCmostrando 11
Phenotype Analysis in Two Families With Otopalatodigital Syndrome Spectrum Disorder Based on FLNA Gene Variants.
Clinical geneticsConcomitant omphalocele, craniorachischisis and ectopic cordis associated with trisomy 18 diagnosed in first trimester.
Taiwanese journal of obstetrics & gynecologyCardiac Surgery in Trisomy 13 and 18: A Guide to Clinical Decision-Making.
Pediatric cardiologyA Male Case of Kagami-Ogata Syndrome Caused by Paternal Unipaternal Disomy 14 as a Result of a Robertsonian Translocation.
Frontiers in pediatricsPPP2R3C gene variants cause syndromic 46,XY gonadal dysgenesis and impaired spermatogenesis in humans.
European journal of endocrinologyPhenotypic spectrum associated with SPECC1L pathogenic variants: new families and critical review of the nosology of Teebi, Opitz GBBB, and Baraitser-Winter syndromes.
European journal of medical geneticsRobinow syndrome: a diagnosis at the fingertips.
Clinical dysmorphologyNonisolated diaphragmatic hernia in Simpson-Golabi-Behmel syndrome.
Prenatal diagnosisThe prevalence of selected major birth defects in the United States.
Seminars in perinatologyThe clinical course of an overgrowth syndrome, from diagnosis in infancy through adulthood: the case of Beckwith-Wiedemann syndrome.
Current problems in pediatric and adolescent health careCyclopia: a rare condition with unusual presentation - a case report.
Clinical medicine insights. PediatricsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de onfalocele familiar com dismorfia facial.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de onfalocele familiar com dismorfia facial
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Phenotype Analysis in Two Families With Otopalatodigital Syndrome Spectrum Disorder Based on FLNA Gene Variants.
- Concomitant omphalocele, craniorachischisis and ectopic cordis associated with trisomy 18 diagnosed in first trimester.
- A Male Case of Kagami-Ogata Syndrome Caused by Paternal Unipaternal Disomy 14 as a Result of a Robertsonian Translocation.
- Cardiac Surgery in Trisomy 13 and 18: A Guide to Clinical Decision-Making.
- PPP2R3C gene variants cause syndromic 46,XY gonadal dysgenesis and impaired spermatogenesis in humans.
- Phenotypic spectrum associated with SPECC1L pathogenic variants: new families and critical review of the nosology of Teebi, Opitz GBBB, and Baraitser-Winter syndromes.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:280403(Orphanet)
- MONDO:0017235(MONDO)
- GARD:21086(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q55786927(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de onfalocele familiar com dismorfia facial
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata