Paraplegia espástica hereditária (PEH) é um grupo de doenças hereditárias cuja principal característica é um distúrbio progressivo da marcha. A doença apresenta-se com rigidez progressiva (espasticidade) e contração nos membros inferiores. A PEH também é conhecida como paraparesia espástica hereditária, paraplegia espástica familiar, doença do assentamento francês, doença de Strumpell ou doença de Strumpell-Lorrain. Os sintomas resultam da disfunção de axônios longos na medula espinhal. As células afetadas são os neurônios motores primários; portanto, a doença é uma doença do neurônio motor superior. A PEH não é uma forma de paralisia cerebral, embora fisicamente possa parecer e se comportar de forma muito semelhante à diplegia espástica.
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome de paraplegia espástica-epilepsia-perturbação do desenvolvimento intelectual é uma condição genética rara que afeta o sistema nervoso. Ela se caracteriza pela combinação de três manifestações principais: paraplegia espástica (rigidez e fraqueza progressiva nas pernas), epilepsia (crises convulsivas) e perturbação do desenvolvimento intelectual (dificuldades de aprendizado e cognição). A doença está registrada no banco de dados OMIM sob o código 182610 e na ontologia Mondo como MONDO:0008439.[1][2][3]
Sinais e sintomas
Os principais sinais e sintomas incluem paraplegia espástica (aumento do tônus muscular e fraqueza nos membros inferiores, que pode levar a dificuldade para caminhar), epilepsia (crises convulsivas de diferentes tipos) e perturbação do desenvolvimento intelectual (atraso no desenvolvimento cognitivo e dificuldades de aprendizado). A apresentação e a gravidade podem variar entre os indivíduos afetados.[1][2]
Causas genéticas
A síndrome é causada por alterações genéticas (mutações) que ainda estão sendo investigadas. Até o momento, não há um gene específico confirmado como causador da condição, mas acredita-se que tenha uma base genética. O padrão de herança não está claramente estabelecido. Para informações atualizadas sobre genes associados, consulte o banco de dados GenCC (search.thegencc.org).[1][2][4]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sintomas característicos (paraplegia espástica, epilepsia e perturbação do desenvolvimento intelectual). Exames complementares, como ressonância magnética do cérebro e eletroencefalograma, podem auxiliar na investigação. O teste genético pode ser útil para confirmar a suspeita diagnóstica, embora nem sempre seja possível identificar a mutação responsável. Consulte um médico geneticista para orientação sobre testes disponíveis.[1][2][4]
Tratamento e manejo
O tratamento é multidisciplinar e focado no controle dos sintomas e na melhora da qualidade de vida. Para a epilepsia, podem ser utilizados medicamentos anticonvulsivantes (como lamotrigina, topiramato, valproato de sódio, entre outros), sempre sob prescrição e acompanhamento médico. A paraplegia espástica pode ser manejada com fisioterapia, terapia ocupacional e, em alguns casos, medicamentos para reduzir a espasticidade. O suporte educacional e psicológico é importante para a perturbação do desenvolvimento intelectual. Não há cura conhecida, e o tratamento é individualizado.[1][2]
Tratamentos citados na literatura
A literatura científica (fonte: PubTator3) menciona associações entre esta síndrome e diversos fármacos, mas estas são apenas correlações observadas em estudos, e não recomendações de tratamento. Os fármacos listados são: aminohydroxybutyric acid, diclofenamide, ethosuximide, felbamate, fosphenytoin, lamotrigine, metharbital, phenacemide, pyridoxal, stiripentol, topiramate, valproic acid, valpromide e zonisamide. O número de publicações para cada fármaco não foi especificado nas fontes disponíveis. Consulte sempre seu médico antes de considerar qualquer medicação.[1][2]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade dos sintomas e a resposta ao tratamento. Com acompanhamento médico adequado e suporte multidisciplinar, muitas pessoas conseguem ter uma qualidade de vida satisfatória, embora possam necessitar de cuidados contínuos. O acompanhamento regular com neurologista, geneticista e equipe de reabilitação é fundamental para otimizar o desenvolvimento e o bem-estar.[1][2]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Paraplegia espástica hereditária (PEH) é um grupo de doenças hereditárias cuja principal característica é um distúrbio progressivo da marcha. A doença apresenta-se com rigidez progressiva (espasticidade) e contração nos membros inferiores. A PEH também é conhecida como paraparesia espástica hereditária, paraplegia espástica familiar, doença do assentamento francês, doença de Strumpell ou doença de Strumpell-Lorrain. Os sintomas resultam da disfunção de axônios longos na medula espinhal. As células afetadas são os neurônios motores primários; portanto, a doença é uma doença do neurônio motor superior. A PEH não é uma forma de paralisia cerebral, embora fisicamente possa parecer e se comportar de forma muito semelhante à diplegia espástica.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome de paraplegia espástica-epilepsia-perturbação do desenvolvimento intelectual é uma condição genética rara que afeta o sistema nervoso. Ela se caracteriza pela combinação de três manifestações principais: paraplegia espástica (rigidez e fraqueza progressiva nas pernas), epilepsia (crises convulsivas) e perturbação do desenvolvimento intelectual (dificuldades de aprendizado e cognição). A doença está registrada no banco de dados OMIM sob o código 182610 e na ontologia Mondo como MONDO:0008439.[1][2][3]
Sinais e sintomas
Os principais sinais e sintomas incluem paraplegia espástica (aumento do tônus muscular e fraqueza nos membros inferiores, que pode levar a dificuldade para caminhar), epilepsia (crises convulsivas de diferentes tipos) e perturbação do desenvolvimento intelectual (atraso no desenvolvimento cognitivo e dificuldades de aprendizado). A apresentação e a gravidade podem variar entre os indivíduos afetados.[1][2]
Causas genéticas
A síndrome é causada por alterações genéticas (mutações) que ainda estão sendo investigadas. Até o momento, não há um gene específico confirmado como causador da condição, mas acredita-se que tenha uma base genética. O padrão de herança não está claramente estabelecido. Para informações atualizadas sobre genes associados, consulte o banco de dados GenCC (search.thegencc.org).[1][2][4]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sintomas característicos (paraplegia espástica, epilepsia e perturbação do desenvolvimento intelectual). Exames complementares, como ressonância magnética do cérebro e eletroencefalograma, podem auxiliar na investigação. O teste genético pode ser útil para confirmar a suspeita diagnóstica, embora nem sempre seja possível identificar a mutação responsável. Consulte um médico geneticista para orientação sobre testes disponíveis.[1][2][4]
Tratamento e manejo
O tratamento é multidisciplinar e focado no controle dos sintomas e na melhora da qualidade de vida. Para a epilepsia, podem ser utilizados medicamentos anticonvulsivantes (como lamotrigina, topiramato, valproato de sódio, entre outros), sempre sob prescrição e acompanhamento médico. A paraplegia espástica pode ser manejada com fisioterapia, terapia ocupacional e, em alguns casos, medicamentos para reduzir a espasticidade. O suporte educacional e psicológico é importante para a perturbação do desenvolvimento intelectual. Não há cura conhecida, e o tratamento é individualizado.[1][2]
Tratamentos citados na literatura
A literatura científica (fonte: PubTator3) menciona associações entre esta síndrome e diversos fármacos, mas estas são apenas correlações observadas em estudos, e não recomendações de tratamento. Os fármacos listados são: aminohydroxybutyric acid, diclofenamide, ethosuximide, felbamate, fosphenytoin, lamotrigine, metharbital, phenacemide, pyridoxal, stiripentol, topiramate, valproic acid, valpromide e zonisamide. O número de publicações para cada fármaco não foi especificado nas fontes disponíveis. Consulte sempre seu médico antes de considerar qualquer medicação.[1][2]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade dos sintomas e a resposta ao tratamento. Com acompanhamento médico adequado e suporte multidisciplinar, muitas pessoas conseguem ter uma qualidade de vida satisfatória, embora possam necessitar de cuidados contínuos. O acompanhamento regular com neurologista, geneticista e equipe de reabilitação é fundamental para otimizar o desenvolvimento e o bem-estar.[1][2]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de paraplegia espástica-epilepsia-perturbação do desenvolvimento intelectual
Centros de Referência SUS
13 centros habilitados pelo SUS para Síndrome de paraplegia espástica-epilepsia-perturbação do desenvolvimento intelectual
Centros para Síndrome de paraplegia espástica-epilepsia-perturbação do desenvolvimento intelectual
Detalhes dos centros
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Expanding the Phenotypic Spectrum of the Recurrent De Novo FBXO31 p.Asp334Asn Variant: Evidence for a Novel Neurodevelopmental Disorder (Kruer Syndrome).
Biallelic loss-of-function variants in FBXO31 cause autosomal-recessive intellectual disability. A recurrent de novo variant, c.1000G>A(p.Asp334Asn), has been described in association with an autosomal-dominant phenotype. To refine this phenotype and its clinical implications, we re-evaluated three published cases and ascertained four additional probands via advocacy networks, GeneMatcher, and clinician referral. Phenotyping included neurologic, behavioral, and dysmorphology assessment. All seven individuals carried the recurrent de novo FBXO31 p.Asp334Asn variant. A core neurodevelopmental profile was observed and included cerebral palsy (mixed hypotonia, spasticity, and dystonia), global developmental delay/intellectual disability, and speech impairment. Neuropsychiatric features were sometimes prominent and included attention-deficit/hyperactivity disorder, anxiety, stereotypies, autistic features, and behavioral dysregulation. Neuroimaging often showed a hypoplastic corpus callosum and posterior-predominant white-matter changes. In one individual, gray matter heterotopias were also observed. A subtle but consistent facial gestalt was noted. Recurrent FBXO31 p.Asp334Asn variants lead to a recognizable neurodevelopmental syndrome. Based on our findings, we recommend including FBXO31 in diagnostic algorithms for cerebral palsy and neurodevelopmental disorders. We propose the descriptive term "autosomal dominant FBXO31-associated neurodevelopmental disorder," and-consistent with the validating laboratory and with support from the FBXO31 Foundation-propose the eponym "Kruer syndrome."
Multisystem manifestations of Sjögren-Larsson syndrome in early childhood and its dental implications.
Sjögren-Larsson syndrome (SLS) constitutes a rare genetic disorder manifesting as a complex neurocutaneous condition characterised by congenital ichthyosis, progressive neurological impairment and intellectual disability. This case report presents an early childhood female patient exhibiting the classic triad of symptoms, along with significant oral complications, including severe dental caries, enamel demineralisation and gingivitis. Molecular genetic testing confirmed a homozygous pathogenic variant in the ALDH3A2 (Aldehyde Dehydrogenase 3 family member 2) gene, establishing the diagnosis. The patient's management encompassed a comprehensive multidisciplinary approach, integrating dental interventions under general anaesthesia, systemic therapies for spasticity and cutaneous manifestations, and regular follow-up care. This case highlights the critical importance of recognising oral manifestations in SLS and emphasises the need for integrated oral healthcare within the broader therapeutic framework for affected individuals.
CTNNB1-related disorders: clinical and radiological contributions from a French cohort.
CTNNB1 monoallelic pathogenic variants account for up to 4% of genetically determined cerebral palsy cases, yet their phenotypic spectrum remains poorly defined. We retrospectively analyzed 25 individuals with pathogenic CTNNB1 variants using medical records and a questionnaire. Data included genetic variants, perinatal history, developmental milestones, behavioral characteristics, head growth, feeding, sleep difficulties, neurological and ophthalmological assessments. Brain MRIs were reviewed by expert neuroradiologists. Twenty-two distinct heterozygous variants were identified. Microcephaly occurred in 16/22 patients. All exhibited global developmental delay, independent walking was achieved at a mean age of 2.1 years, with regression in 4/16 independent walkers. Behavioral disorders were frequent, as were oral sensorimotor disorders (21/25) and sleep disturbances (13/21). Lower limb hypertonia was present in 22/25 patients [spastic (8) and/or dystonic (11)]. Unstable gait were common among ambulatory patients. Exaggerated startle reactions, often since birth, were reported in 16/21. Exudative vitreoretinopathy was identified in 3/5 patients with retinal angiography. Brain MRI (19 patients) showed: thickening of anterior commissure (8), frontal lobe hypoplasia (9), widening of superior vermian sulci (10) and corpus callosum anomalies (7). This study broadens the spectrum of CTNNB1-related syndrome, reporting a complex motor phenotype combining (i) gait disturbances related to dystonic or non-dystonic hypertonia and unsteadiness, sometimes associated to dystonia in other body parts (ii) possible deterioration of motor achievements over the course of the disease (iii) an exaggerated startle reflex. New non-specific brain anomalies are precisely described. Our work underscores the need for registries and longitudinal studies to refine characterization and guide future therapies. PTS-related tetrahydrobiopterin deficiency (PTPSD) results in a lack of tetrahydropterin, an important cofactor for phenylalanine hydroxylase (PAH), tyrosine hydroxylase, and tryptophan hydroxylase. Deficiency can thus lead to neurotransmitter and neuropsychiatric disorders. The clinical spectrum of PTPSD is broad and differs according to age of onset, severity of disease, and whether preventative therapies were initiated and maintained from an early age. In the severe form, clinical symptoms may become apparent in the neonatal period and can include hypotonia, movement disorders, abnormal eye movements, autonomic dysregulation, and impaired development. Without treatment, developmental delays become more marked. Neurologic symptoms (dysarthria, dystonia, tremors, abnormal gait, parkinsonism, oculogyric crises, motor tics) may be ameliorated by treatment with sapropterin dihydrochloride and neurotransmitter precursors. Other features of the condition can include psychiatric comorbidities (ADHD, anxiety, depression), infant feeding difficulties leading to early growth failure, hyperprolactinemia, growth hormone deficiency, sleep issues, and autonomic dysfunction; many of these features can be ameliorated by appropriate treatment. In treated individuals, development often improves during adolescence, with many adults having a normal IQ level. In the mild (peripheral) form, affected individuals are usually asymptomatic apart from an increase in phenylalanine (Phe) levels. Some remain asymptomatic. However, with time, some have mild developmental delays and can develop deficiency of neurotransmitter production, such that treatment of some asymptomatic individuals may be required. The biochemical diagnosis of PTPSD is established in a proband with confirmed hyperphenylalaninemia, elevated neopterin levels, reduced biopterin levels, and a decreased biopterin-to-neopterin ratio in urine or dried blood spots (DBS) and normal dihydropteridine reductase (DHPR) activity in DBS. The molecular diagnosis of PTPSD is established in a proband by identification of biallelic pathogenic (or likely pathogenic) variants in PTS by molecular genetic testing. Targeted therapies: Immediate therapy with sapropterin (tetrahydrobiopterin dihydrochloride; BH4), a cofactor/cosubstrate of PAH, is recommended to reduce blood Phe concentrations in individuals with hyperphenylalaninemia. If sapropterin is not available, dietary Phe restriction should be implemented. Because sapropterin has limited access to the central nervous system (CNS), or rather, this access is only achieved at high doses, therapy with sapropterin does not normalize the activity of tyrosine or tryptophan hydroxylase in people with PTPSD. Additional treatment strategies are necessary for long-term management and may include the use of neurotransmitter precursors (levodopa plus decarboxylase inhibitor (DCI), i.e., carbidopa or benserazide), 5-hydroxytryptophan, and/or dopamine (rotigotine patch, pramipexole) and/or serotonin agonists, or other medications (MAO inhibitors such as selegiline) to address specific neurotransmitter deficiencies and maintain optimal neurologic function. Supportive care: Optimization of dosage and intervals of levodopa/DCI in those with abnormal movements/parkinsonism; growth hormone supplementation and/or optimization of neurotransmitter precursor therapy for growth hormone deficiency; optimization of neurotransmitter precursor therapy for recurrent hyperthermia; anticholinergic treatment may be considered for hypersalivation; standard treatment for developmental delay, spasticity, epilepsy, sleep disorders, and decreased bone mineral density. Biochemical surveillance: Routine Phe monitoring in infants (age <1 year) weekly until normalized and then every three to six months once levels normalize; every six months in children younger than age 12 years; and every six to 12 months in adolescents and adults; the Phe target ranges correspond to those of PAH deficiency. Prolactin level at each visit. Routine clinical visits with a metabolic specialist (and metabolic dietician if on Phe-restricted diet) every one to three months in infants (age <1 year), every three to six months between ages one and seven years, and every six to 12 months in those age eight years and older. General surveillance: At each visit, measure growth parameters and evaluate nutritional status; asses for new neurologic manifestations (changes in tone, seizures, movement disorders); monitor developmental progress and assess educational needs; monitor for behavioral issues (anxiety, ADHD, emotional dysregulation, depression, aggression); and assess for signs and symptoms of sleep disorders. At ages two, six, 12, and 18 years, consider neuropsychological evaluation. In adulthood, periodic parathormone levels and DXA scan. As needed, consider EEG to differentiate from movement disorder seizures. Agents/circumstances to avoid: Persons with PTPSD on Phe-reduced diet should either avoid products containing aspartame or calculate total intake of Phe when using such products and adapt diet components accordingly. Evaluation of relatives at risk: If prenatal genetic testing has not been performed, each at-risk newborn sib should be evaluated immediately (at or just after 24 hours) after birth for PTPSD using measurement of blood Phe concentration to allow for earliest possible diagnosis and treatment. If older sibs have not undergone NBS or genetic testing for the known familial pathogenic variants in PTS, measure blood Phe concentrations to clarify their disease status. Pregnancy management: Women with PTPSD who have received appropriate treatment throughout childhood and adolescence and during pregnancy may have offspring with normal intellectual and behavioral development, particularly if levels of Phe are kept in the normal range during pregnancy. Intensive clinical and biochemical supervision by a multidisciplinary team before, during, and after pregnancy in a woman with PTPSD is essential to control the symptoms of the disease, adjust the treatment if needed, and monitor the development of the fetus. If the affected woman has elevated blood Phe concentrations during pregnancy, the fetus is at high risk for maternal phenylketonuria (MPKU) syndrome (reported specifically in women who have PAH deficiency as the primary cause of their elevated Phe levels), including malformations and intellectual disability, since Phe is a potent teratogen. PTPSD is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a PTS pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of inheriting neither of the familial PTS pathogenic variants. Children born of one parent with PTPSD and one parent with two normal PTS alleles are obligate heterozygotes. If the mother is the affected parent, MPKU syndrome is a critical issue. Females with PTPSD should receive counseling regarding the teratogenic effects of elevated maternal plasma Phe concentration (i.e., MPKU syndrome) when they reach childbearing age. Once the PTS pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives and prenatal/preimplantation genetic testing for PTPSD are possible.
Neuroradiological Phenotype Expansion of the Siddiqi Syndrome: A Case Report.
Siddiqi syndrome is a rare autosomal recessive deafness-dystonia disorder caused by pathogenic variants in the FITM2 gene. To date, only 5 unrelated families have been reported in the literature carrying loss-of-function variants in FIMT2 gene. In this report, we describe a 29-year-old woman with compound heterozygous novel variants identified by trio-based exome sequencing. She carries the paternally inherited delins variant c.158_161delinsTCAT, p.(Arg53_Asn54delinsLeuIle) and the maternally inherited frameshift variant c.567del, p.(Thr190ProfsTer9) in FITM2 gene. The patient exhibits the main features of the disease, including deafness, intellectual disability, regression of motor skills and poor overall growth. Additionally, she presents with spastic paraplegia which supports recent phenotypic expansion. We describe for the first time, novel brain magnetic resonance imaging signal alterations, not previously associated with this disorder. These neuroimaging findings may provide new insights into the neurological manifestations of Siddiqi syndrome. This case expands the phenotypic and molecular spectrum of FITM2 associated disease and emphasizes the adult-features of this syndrome. [Image: see text]
Bi-allelic variants in FSD1L cause a neurodevelopmental disorder overlapping with L1 syndrome.
Disruption of the complex processes underlying central nervous system development leads to a broad spectrum of brain malformations and neurodevelopmental disorders, often with a genetic cause. Here, we report bi-allelic pathogenic variants in fibronectin type III and SPRY domain-containing 1-like (FSD1L), encoding a protein of unknown function, in eleven individuals, including five fetuses from six unrelated families. The phenotype ranges from severe hydrocephalus, corpus callosum agenesis, and absent pyramid decussation to a neurodevelopmental syndrome characterized by severe intellectual disability, spastic tetraparesis, reduced vision, and epilepsy, associated with corpus callosum agenesis/hypoplasia, mild ventricular dilation, optic nerve hypoplasia, and white matter reduction. This phenotype closely resembles that observed in L1 syndrome, caused by pathogenic variants in L1CAM, encoding a neural adhesion molecule. The knockdown of Fsd1l in mouse embryos recapitulated the ventricular dilation observed in affected fetuses. Immunohistochemical studies in human control fetuses revealed that FSD1L localized to neurons with commissural fate and projection neurons during human development. Induced pluripotent stem cell (iPSC)-derived neural progenitor cells from affected individuals failed to differentiate into premature neurons and to properly form neurospheres while undergoing increased cell death. In neural progenitors, FSD1L localized with microtubules of the mitotic spindle during M phase and to the transition zone and along the axoneme of the primary cilium during interphase. In line with this, fibroblasts from affected individuals exhibited marked alterations of the mitotic spindle and reduced ciliogenesis and ciliary length compared to control cells. Our findings define FSD1L as a microtubule-associated protein implicated in neuronal differentiation, axon guidance, and fasciculation.
Publicações recentes
Bi-allelic variants in FSD1L cause a neurodevelopmental disorder overlapping with L1 syndrome.
Biallelic TTBK1 variant causes a severe syndromic neurodevelopmental disorder: clinical and genetic insights from two siblings.
The first description of CTNNB1 syndrome in the Tunisian population: clinical investigation, molecular docking and molecular dynamics simulation of β-catenin/E-cadherin complex.
Gait abnormalities in children with FOXP1 syndrome: A case series.
New Insights Into TRMT10A Syndrome: Case Report and Literature Review.
📚 EuropePMCmostrando 199
Expanding the Phenotypic Spectrum of the Recurrent De Novo FBXO31 p.Asp334Asn Variant: Evidence for a Novel Neurodevelopmental Disorder (Kruer Syndrome).
Clinical geneticsMultisystem manifestations of Sjögren-Larsson syndrome in early childhood and its dental implications.
BMJ case reportsCTNNB1-related disorders: clinical and radiological contributions from a French cohort.
Frontiers in neurologyNeuroradiological Phenotype Expansion of the Siddiqi Syndrome: A Case Report.
Cellular and molecular neurobiologyBi-allelic variants in FSD1L cause a neurodevelopmental disorder overlapping with L1 syndrome.
American journal of human geneticsMorbidity and Mortality Among People Living With HTLV-1: A 30-Year Retrospective Analysis in a Brazilian Cohort.
Journal of medical virologyPain in multiple sclerosis: clinical phenotypes and therapeutic strategies - a narrative review.
Pain reportsExpanding the clinical and immunological phenotypes of COPB1 deficiency.
Frontiers in immunologyEarly Recognition and Intervention for Poststroke Spasticity: A Scientific Statement From the American Heart Association.
StrokeCoffin-Lowry syndrome: a systematic review of RPS6KA3 confirmed cases and implications for diagnosis and counseling.
Frontiers in geneticsExpanding the neurological spectrum of HTLV-1 beyond HAM/TSP: a contemporary perspective.
Lancet regional health. AmericasBiallelic TTBK1 variant causes a severe syndromic neurodevelopmental disorder: clinical and genetic insights from two siblings.
Journal of medical geneticsA proof-of-concept study on the effectiveness of botulinum toxin on spasticity plus syndrome in multiple sclerosis.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyLennox-Gastaut syndrome: Comorbidities and clinical implications.
Seminars in pediatric neurologyDeciphering Spastic Ataxia: Clinical and Genetic Profiles.
Neurology. GeneticsNeurodegeneration With Brain Iron Accumulation and Ferroptosis Disorders in Children and Adults: An Imaging Review.
Journal of neuroimaging : official journal of the American Society of NeuroimagingLeukoencephalopathy, brain calcifications, and cysts (LCC): Two unique cases.
Rare (Amsterdam, Netherlands)Two Chinese patients with Basilicata-Akhtar syndrome caused by novel MSL3 variants: a case report and literature review.
Translational pediatricsAltered Dopamine Metabolism and Response to Treatment with Levodopa/Carbidopa in MCT8 Deficiency.
Movement disorders : official journal of the Movement Disorder SocietyThe first description of CTNNB1 syndrome in the Tunisian population: clinical investigation, molecular docking and molecular dynamics simulation of β-catenin/E-cadherin complex.
Molecular biology reportsATP6AP2-Related Disease Caused by Splicing Defects: Abnormal Glycosylation and the First Affected Female.
Journal of inherited metabolic diseaseBi-allelic variants in the ribosomal protein RPS6KC1 cause a complex neurodevelopmental disorder.
American journal of human geneticsSelective peripheral neurotomy for multifocal spasticity: Two-dimensional operative video.
Surgical neurology internationalBiallelic NDUFA9 variants cause a progressive neurodevelopmental disorder with prominent dystonia and mitochondrial complex I deficiency.
Brain communicationsPeripheral neuromodulation in spasticity-plus syndrome: effects of pulsed radiofrequency on tonic-painful disorders in multiple sclerosis.
Frontiers in neurologyDupilumab Reduces Pruritus in Twins With Sjögren-Larsson Syndrome.
Pediatric dermatologyKohlschütter-Tönz Syndrome: A Rare Clinical Entity with Amelogenesis Imperfecta in Two Siblings, Dental Management and Scoping Review.
Turkish archives of pediatricsThe efficacy and safety of the ketogenic diet in infantile epileptic spasm syndrome: a meta-analysis.
SeizureA Case of Infantile Epileptic Spasms Syndrome with the SPTBN1 Mutation and Review of βII-Spectrin Variants.
GenesCase report of paroxysmal dystonia in a child with KBG syndrome: Expansion of the phenotype and utility of whole exome sequencing.
MedicineMaternal uniparental isodisomy in a patient with autosomal recessive spastic paraplegia type 20.
GeneGait abnormalities in children with FOXP1 syndrome: A case series.
Journal of pediatric rehabilitation medicineGenome Sequencing Uncovers Additional Findings in Phelan-McDermid Syndrome.
American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric GeneticsA Report of a Child with SEC31A-Related Neurodevelopmental Disorder.
International journal of molecular sciencesFrench guidelines for the diagnosis and management of pure hereditary spastic paraplegia.
Revue neurologiqueFrench protocol for diagnosis and management of type 1 interferonopathies.
La Revue de medecine interneFurther Delineation of the AUTS2 HX Repeat Domain-Related Phenotype.
American journal of medical genetics. Part AExpanding the spectrum of ATP8A2 mutations: a new splicing variant and systematic review of CAMRQ4 syndrome.
Molecular biology reportsArginase 1 deficiency: a treatable form of spastic paraplegia.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologySensory Dysfunction in ALS and Other Motor Neuron Diseases: Clinical Relevance, Histopathology, Neurophysiology, and Insights from Neuroimaging.
BiomedicinesThe implications of hyperekplexia on children's quality of life: a report on two cases.
Revista paulista de pediatria : orgao oficial da Sociedade de Pediatria de Sao PauloIdentification of a novel non-coding deletion in Allan-Herndon-Dudley syndrome by long-read HiFi genome sequencing.
BMC medical genomicsDe Novo Deletion in the 12q24.23q24.31 Chromosomal Region Causing a Neurodevelopmental Syndrome in a Female Saudi Patient: A Case Report.
CureusOutpatient Management of Clinical Comorbidities in Children With Cerebral Palsy in Low- and Middle-Income Countries.
Child: care, health and developmentAdult Leigh Syndrome Associated with the m.15635T>C Mitochondrial DNA Variant Affecting the Cytochrome b (MT-CYB) Gene.
International journal of molecular sciencesKidins220-deficient hydrocephalus mice exhibit altered glial phenotypes and AQP4 differential regulation in the retina and optic nerve, with preserved retinal ganglion cell survival.
Fluids and barriers of the CNSAmbulatory children with spastic cerebral palsy have smaller bone area and deficits in trabecular microarchitecture.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchWorsening spasticity due to catheter breakage during intrathecal baclofen therapy: a case report.
Journal of medical case reportsIntrathecal Baclofen Pump Wean and Risk of Exposure in a Large Total Body Surface Area Burn Wound Patient: A Case Report.
Journal of burn care & research : official publication of the American Burn AssociationSciatic nerve analysis in thyroid hormone transporters Mct8 and Oatp1c1 knockout mice.
European thyroid journalCerebrotendinous xanthomatosis: A complex interplay between a clinically and genetically heterogeneous condition.
European journal of neurologyFrom spastic paraplegia to infantile neurodegenerative disorder: Expanding the phenotypic spectrum associated with biallelic SPAST variants.
European journal of neurologyCHD3-related Snijders Blok-Campeau syndrome with Spastic Paraplegia, Ataxia, and Situs Inversus.
European journal of medical genetics[Current state of algology - pain medicine - in the Russian Federation].
Zhurnal voprosy neirokhirurgii imeni N. N. BurdenkoAccumulation of ether phospholipids in induced pluripotent stem cells and oligodendrocyte-lineage cells established from patients with Sjögren-Larsson syndrome.
Congenital anomaliesCervical myelopathy mistaken for complex regional pain syndrome: A case report.
MedicineNew Insights Into TRMT10A Syndrome: Case Report and Literature Review.
American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric GeneticsSynaptic dynamics linked to widespread elevation of H-reflex before peripheral denervation in amyotrophic lateral sclerosis.
Journal of neurophysiologyParticularities of spasticity in myelomeningocele patients.
Child's nervous system : ChNS : official journal of the International Society for Pediatric NeurosurgeryPrenatal Diagnosis of a KIDINS220 De Novo Heterozygous Variant in a Fetus With a Complex CNS Anomaly.
Prenatal diagnosisAtlantoaxial Instability due to Os Odontoideum in a Child with Christianson Syndrome.
Molecular syndromologyThe L1CAM SAX-7 is an antagonistic modulator of Erk Signaling.
bioRxiv : the preprint server for biologyA Novel KIDINS220 Pathogenic Variant Associated with the Syndromic Spastic Paraplegia SINO: An Expansion of the Brain Malformation Spectrum and a Literature Review.
GenesBiallelic PI4KA Mutations Disrupt B-Cell Metabolism and Cause B-Cell Lymphopenia and Hypogammaglobulinemia.
Journal of clinical immunologyNovel autosomal recessive SINO syndrome-associated KIDINS220 variants provide insight into the genotype-phenotype correlation.
HeliyonCombined generalized and focal epilepsy with reflex features in Adaptor protein complex 4-associated hereditary spastic paraplegias: A cohort observational study.
SeizureA case report of spastic diplegic cerebral palsy in a late preterm child with hypoplastic left heart syndrome.
Translational pediatricsSuccessful Electroconvulsive Therapy in Aicardi-Goutières Syndrome Presenting Psychiatric Symptoms: An Unprecedented Clinical Case.
The journal of ECTPure Hereditary Spastic Paraplegia in a Patient With a Novel Heterozygous KIDINS220 Gene Mutation.
CureusA novel missense variant in the ATPase domain of ATP8A2 and review of phenotypic variability of ATP8A2-related disorders caused by missense changes.
NeurogeneticsRefining the phenotype of SINO syndrome: A comprehensive cohort report of 14 novel cases.
Genetics in medicine : official journal of the American College of Medical GeneticsThe Effectiveness of Combining Botulinum Toxin Type A and Therapeutic Exercise in Treating Spasticity in a Patient with Complicated Stiff-Person Syndrome: A Case Report.
Diseases (Basel, Switzerland)De novo and inherited monoallelic variants in TUBA4A cause ataxia and spasticity.
Brain : a journal of neurology[Intrathecal baclofen therapy: a 30-year experience].
Zhurnal voprosy neirokhirurgii imeni N. N. BurdenkoExpanding the phenotype of Harel-Yoon syndrome: A case report suggesting a genotype/phenotype correlation.
American journal of medical genetics. Part AA Missense Variant in HACE1 Is Associated with Intellectual Disability, Epilepsy, Spasticity, and Psychomotor Impairment in a Pakistani Kindred.
GenesElectroacupuncture stimulation modulates functional brain connectivity in the treatment of pediatric cerebral palsy: a case report.
Frontiers in psychiatryA novel AP1S2 variant causing leaky splicing in X-linked intellectual disability: Further delineation and intrafamilial variability.
American journal of medical genetics. Part AAn Ultra-Rare Mixed Phenotype with Combined AP-4 and ERF Mutations: The First Report in a Pediatric Patient and a Literature Review.
GenesUtilizing combination intrathecal baclofen and analgesic medication to manage spasticity and pain in patients with pediatric-onset disability: Case series.
Journal of pediatric rehabilitation medicineSingle Nucleotide Polymorphism in Cell Adhesion Molecule L1 Affects Learning and Memory in a Mouse Model of Traumatic Brain Injury.
International journal of molecular sciencesPediatric Spastic Wrist Contractures Can Be Well Managed With Wrist Arthrodesis.
Journal of pediatric orthopedicsType 1 early infantile epileptic encephalopathy: A case report and literature review.
Molecular genetics & genomic medicineAlterations in KIDINS220/ARMS Expression Impact Sensory Processing and Social Behavior in Adult Mice.
International journal of molecular sciencesFlexor Pronator Slide Under Local Anesthesia without a Tourniquet for Non-Ischemic Contractures of the Forearm.
JBJS essential surgical techniquesFirst report of Ageratum yellow vein virus infecting papaya in Lampung, Indonesia.
Molecular biology reportsNovel loss-of-function variants expand ABCC9-related intellectual disability and myopathy syndrome.
Brain : a journal of neurologyMedical comorbidities in patients with prolonged disorder of consciousness: A narrative review.
NeuroRehabilitationGeneration of three induced pluripotent stem cell lines from individuals with Aicardi-Goutières syndrome caused by a c.3019G>A (p.G1007R) autosomal dominant pathogenic variant in ADAR1.
Stem cell researchThe Rogdi knockout mouse is a model for Kohlschütter-Tönz syndrome.
Scientific reportsNovel Homozygous Variants of SLC13A5 Expand the Functional Heterogeneity of a Homogeneous Syndrome of Early Infantile Epileptic Encephalopathy.
Pediatric neurologyReport and Abstracts of the 15th Congress of the Mediterranean Forum of Physical and Rehabilitation Medicine: Rome, July 6-8, 2023.
European journal of translational myology[The use of botulinum toxin type A in symptomatic therapy and medical rehabilitation of patients with multiple sclerosis].
Zhurnal nevrologii i psikhiatrii imeni S.S. KorsakovaBi-allelic ACBD6 variants lead to a neurodevelopmental syndrome with progressive and complex movement disorders.
Brain : a journal of neurologyMethyl-CpG-Binding protein 2 duplication syndrome in a Chinese patient: A case report and review of the literature.
World journal of clinical casesPathophysiology of cervical myelopathy (Review).
Biomedical reportsPanoramic variation analysis of a family with neurodevelopmental disorders caused by biallelic loss-of-function variants in TMEM141, DDHD2, and LHFPL5.
Frontiers of medicineIntranasal human-recombinant NGF administration improves outcome in children with post-traumatic unresponsive wakefulness syndrome.
Biology directIdentification of novel homozygous variants in FOXE3 and AP4M1 underlying congenital syndromic anophthalmia and microphthalmia.
The journal of gene medicineTowards Preclinical Validation of Arbaclofen (R-baclofen) Treatment for 16p11.2 Deletion Syndrome.
bioRxiv : the preprint server for biologySERAC1 Deficiency- A New Phenotype.
Endocrine, metabolic & immune disorders drug targetsCorticospinal tract: a new hope for the treatment of post-stroke spasticity.
Acta neurologica BelgicaIntellectual Disability and Behavioral Deficits Linked to CYFIP1 Missense Variants Disrupting Actin Polymerization.
Biological psychiatryEpigenetic regulation of autophagy-related genes: Implications for neurodevelopmental disorders.
AutophagyA biallelic loss-of-function variant in TMEM147 causes profound intellectual disability and spasticity.
NeurogeneticsAicardi syndrome in a Nigerian female child: A case report and literature review of a rare neuro-developmental disorder from North-Western Nigeria.
Journal of the National Medical AssociationTropical spastic paraparesis.
Handbook of clinical neurologyRepetitive Sleep Starts in Allan-Herndon-Dudley Syndrome.
Pediatric neurologyBiallelic MED27 variants lead to variable ponto-cerebello-lental degeneration with movement disorders.
Brain : a journal of neurologyOccupational Therapy Intervention in the Child with Leukodystrophy: Case Report.
Children (Basel, Switzerland)Congenital heart defects in CTNNB1 syndrome: Raising clinical awareness.
Clinical geneticsJuvenile Dermatomyositis and Infantile Cerebral Palsy: Aicardi-Gouteres Syndrome, Type 5, with a Novel Mutation in SAMHD1-A Case Report.
BiomedicinesUntargeted Metabolomic Analysis of Sjögren-Larsson Syndrome Reveals a Distinctive Pattern of Multiple Disrupted Biochemical Pathways.
MetabolitesEpileptic encephalopathy and amelogenesis imperfecta: What about KohlschüttereTönz syndrome? Case report and literature review.
Special care in dentistry : official publication of the American Association of Hospital Dentists, the Academy of Dentistry for the Handicapped, and the American Society for Geriatric DentistryExpanding the genetic spectrum of ALKU syndrome: Compound heterozygosity for two deleterious variants in SMG8 gene.
American journal of medical genetics. Part AExome sequencing identifies a novel pathogenic variant in RAB3GAP1 causing Warburg Micro syndrome in a Pakistani family.
International journal of developmental neuroscience : the official journal of the International Society for Developmental NeuroscienceAdolescent/Adult-Onset Leukodystrophy with MTHFR Deficiency - A Treatable Cause.
Neurology IndiaEarly versus late injections of Botulinumtoxin type A in post-stroke spastic movement disorder: A literature review.
Toxicon : official journal of the International Society on ToxinologyThe Gross, Fine, and Oral Motor Functions in a Patient with Megalencephalic Leukoencephalopathy with Subcortical Cyst: A Case Report.
Iranian journal of child neurologyCation leak through the ATP1A3 pump causes spasticity and intellectual disability.
Brain : a journal of neurologyQuantitative measures of motor development in Angelman syndrome.
American journal of medical genetics. Part AMECP2 duplication syndrome initially misdiagnosed as cerebral palsy: a case report.
The Journal of international medical researchSjögren-Larsson Syndrome: A Rare Presentation With Developmental Delay.
CureusAdar-associated Aicardi Goutières syndrome in a child with bilateral striatal necrosis and recurrent episodes of transaminitis.
BMJ case reportsDuplication within two regions distal to MECP2: clinical similarity with MECP2 duplication syndrome.
BMC medical genomicsNovel, homozygous RAB3GAP1 c.2606 + 1G>A, p.Glu830ValfsTer9 variant and chromosome 3q29 duplication in a Turkish individual with Warburg micro syndrome.
Clinical dysmorphologyThe Mitochondrial tRNASer(UCN) Gene: A Novel m.7484A>G Mutation Associated with Mitochondrial Encephalomyopathy and Literature Review.
Life (Basel, Switzerland)Moyamoya syndrome secondary to mitochondrial disease in a patient with partial trisomy 13q14 and 13q31: A novel case report and literature review.
HeliyonManifestations of Intellectual Disability, Dystonia, and Parkinson's Disease in an Adult Patient with ARX Gene Mutation c.558_560dup p.(Pro187dup).
Case reports in geneticsSpasticity evaluation and management tools.
Muscle & nerveTELO2-related syndrome (You-Hoover-Fong syndrome): Description of 14 new affected individuals and review of the literature.
American journal of medical genetics. Part AThe clinical and genetic spectrum of autosomal-recessive TOR1A-related disorders.
Brain : a journal of neurologyAp4b1-knockout mouse model of hereditary spastic paraplegia type 47 displays motor dysfunction, aberrant brain morphology and ATG9A mislocalization.
Brain communicationsEstablishment of a flow cytometry screening method for patients with glucose transporter 1 deficiency syndrome.
Molecular genetics and metabolism reportsPre- and Postnatal Characterization of Autosomal Recessive KIDINS220-Associated Ventriculomegaly.
Molecular syndromologyCatatonia and Neuroleptic Malignant Syndrome in Patients With Cerebral Palsy: Two Case Reports and a Systematic Review of the Literature.
Journal of the Academy of Consultation-Liaison PsychiatryWhole-Exome Sequencing and Copy Number Analysis in a Patient with Warburg Micro Syndrome.
GenesPreviously Undescribed Gross HACE1 Deletions as a Cause of Autosomal Recessive Spastic Paraplegia.
GenesEffects of L1 adhesion molecule agonistic mimetics on signal transduction in neuronal functions.
Biochemical and biophysical research communicationsBlended Phenotype of Prader-Willi Syndrome and HSP-SPG11 Caused by Maternal Uniparental Isodisomy.
Neurology. GeneticsAutosomal Recessive Spastic Ataxia of Charlevoix-Saguenay due to Novel Mutations in the SACS Gene.
Journal of investigative medicine high impact case reportsPutative founder effect of Arg338* AP4M1 (SPG50) variant causing severe intellectual disability, epilepsy and spastic paraplegia: Report of three families.
Clinical geneticsClinical and genetic analyses in syndromic intellectual disability with primary microcephaly reveal biallelic and de novo variants in patients with parental consanguinity.
Genes & genomicsTCEAL1 loss-of-function results in an X-linked dominant neurodevelopmental syndrome and drives the neurological disease trait in Xq22.2 deletions.
American journal of human geneticsBardet-Biedl Syndrome: A Rare Case From Ophthalmology Perspective.
CureusBrazilian practice guidelines for stroke rehabilitation: Part II.
Arquivos de neuro-psiquiatriaA Novel de novo KIF1A Mutation in a Patient with Ataxia, Intellectual Disability and Mild Foot Deformity.
Cerebellum (London, England)Spastic paraplegia 51: phenotypic spectrum related to novel homozygous AP4E1 mutation.
Journal of geneticsAn association study of IL2RA polymorphisms with cerebral palsy in a Chinese population.
BMC medical genomicsGenetic and clinical characteristics of 24 mainland Chinese patients with CTNNB1 loss-of-function variants.
Molecular genetics & genomic medicineCase report: Huppke-Brendel syndrome in an adult, mistaken for and treated as Wilson disease for 25 years.
Frontiers in neurologyEarly-Onset and Severe Complex Hereditary Spastic Paraplegia Caused by De Novo Variants in SPAST.
Movement disorders : official journal of the Movement Disorder SocietyGenomic and phenotypic characterization of 404 individuals with neurodevelopmental disorders caused by CTNNB1 variants.
Genetics in medicine : official journal of the American College of Medical GeneticsMultiple sclerosis and migraine: Links, management and implications.
Multiple sclerosis and related disordersRefractory status epilepticus with fever due to mumps vaccine-induced encephalitis caused secondary encephalopathy mimicking acute encephalopathy with biphasic seizures and late reduced diffusion.
Brain & developmentAP-4 regulates neuronal lysosome composition, function, and transport via regulating export of critical lysosome receptor proteins at the trans-Golgi network.
Molecular biology of the cell[Nociceptive and mixed pains in patients with multiple sclerosis].
Zhurnal nevrologii i psikhiatrii imeni S.S. KorsakovaNovel CTNNB1 variant leading to neurodevelopmental disorder with spastic diplegia and visual defects plus peripheral neuropathy: A case report.
American journal of medical genetics. Part ASeropositive Neuromyelitis Optica in a Case of Undiagnosed Ankylosing Spondylitis: A Neuro-Rheumatological Conundrum.
Qatar medical journalASXL3 De Novo Variant-Related Neurodevelopmental Disorder Presenting as Dystonic Cerebral Palsy.
NeuropediatricsPSMC1 variant causes a novel neurological syndrome.
Clinical geneticsRetinal Detachment Present at Birth in an Infant With a Novel CTNNB1 Mutation.
Ophthalmic surgery, lasers & imaging retinaNovel EPG5 Mutation Associated with Vici Syndrome Gene.
Case reports in geneticsA homozygous Y443C variant in the RNPC3 is associated with severe syndromic congenital hypopituitarism and diffuse brain atrophy.
American journal of medical genetics. Part AAnalysis of L1CAM gene mutation and imaging appearance in three Chinese families with L1 syndrome: Three case reports.
Molecular genetics & genomic medicineMutations in TAF8 cause a neurodegenerative disorder.
Brain : a journal of neurology[Restoration of hand function in patients with hemiparesis using mirror therapy in combination with myofascial stretching and postisometric relaxation].
Voprosy kurortologii, fizioterapii, i lechebnoi fizicheskoi kulturyQuantitative Three-Dimensional Gait Evaluation in Patients With Glucose Transporter 1 Deficiency Syndrome.
Pediatric neurologyAn Observational Cross-Sectional Study of Gender and Disability as Determinants of Person-Centered Medicine in Botulinum Neurotoxin Treatment of Upper Motoneuron Syndrome.
ToxinsAn ultra-long new onset refractory status epilepticus: Winning the battle but losing the war?
Epilepsy & behavior reports[Connotation of Baimai disease and analysis of compatibility and usage of Baimai Ointment: based on Tibetan medicine theory].
Zhongguo Zhong yao za zhi = Zhongguo zhongyao zazhi = China journal of Chinese materia medicaMolecular Characterization of Portuguese Patients with Hereditary Cerebellar Ataxia.
CellsNovel CAPN1 missense variants in complex hereditary spastic paraplegia with early-onset psychosis.
Annals of clinical and translational neurologyGait as a quantitative translational outcome measure in Angelman syndrome.
Autism research : official journal of the International Society for Autism ResearchMoyamoya syndrome in a young person with Down syndrome: diagnostic and therapeutic considerations.
BMJ case reportsMotor and behavioral phenotype of Dravet syndrome in adulthood.
Epilepsy & behavior : E&BPRUNE1 c.933G>A synonymous variant induces exon 7 skipping, disrupts the DHHA2 domain, and leads to an atypical NMIHBA syndrome presentation: Case report and review of the literature.
American journal of medical genetics. Part AMolybdenum cofactor deficiency: A natural history.
Journal of inherited metabolic diseaseDe Novo and Dominantly Inherited SPTAN1 Mutations Cause Spastic Paraplegia and Cerebellar Ataxia.
Movement disorders : official journal of the Movement Disorder SocietyGenetic diagnosis in Sudanese and Tunisian families with syndromic intellectual disability through exome sequencing.
Annals of human geneticsDe Novo ATP1A1 Variants in an Early-Onset Complex Neurodevelopmental Syndrome.
NeurologyBi-allelic variants in neuronal cell adhesion molecule cause a neurodevelopmental disorder characterized by developmental delay, hypotonia, neuropathy/spasticity.
American journal of human geneticsRhomboencephalosynapsis: Review of the Literature.
World neurosurgeryL1 Syndrome Prenatal Diagnosis Supplemented by Functional Analysis of One L1CAM Gene Missense Variant.
Reproductive sciences (Thousand Oaks, Calif.)Intravenous ketogenic diet therapy for neonatal-onset pyruvate dehydrogenase complex deficiency.
Brain & development[Clinical phenotype and genetic analysis of MECP2 duplication syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsDevelopment and Internal Validation of a Disability Algorithm for Multiple Sclerosis in Administrative Data.
Frontiers in neurologyA Diagnostic Approach to Spastic ataxia Syndromes.
Cerebellum (London, England)A novel homozygous synonymous variant further expands the phenotypic spectrum of POLR3A-related pathologies.
American journal of medical genetics. Part AFunctions of the (pro)renin receptor (Atp6ap2) at molecular and system levels: pathological implications in hypertension, renal and brain development, inflammation, and fibrosis.
Pharmacological researchDysfunctional Homozygous VRK1-D263G Variant Impairs the Assembly of Cajal Bodies and DNA Damage Response in Hereditary Spastic Paraplegia.
Neurology. GeneticsElectroclinical Features in MECP2 Duplication Syndrome: Pediatric Case Series.
Journal of child neurologyChédiak-Higashi syndrome presenting as a hereditary spastic paraplegia.
Journal of human geneticsIntrathecal baclofen therapy for severe spasticity in an adult with tethered cord syndrome: a case report.
Journal of medical case reportsFunctional and Clinical Outcomes of Combined Simultaneous Bilateral Anterior Distal Femoral Plate Hemiepiphysiodesis and Hamstrings Release in Management of Knee Flexion Contractures in Children With Neuromuscular Disorders.
Journal of pediatric orthopedicsHip Displacement in MECP2 Disorders: Prevalence and Risk Factors.
Journal of pediatric orthopedicsRe-analysis of whole-exome sequencing data reveals a novel splicing variant in the SLC2A1 in a patient with GLUT1 Deficiency Syndrome 1 accompanied by hemangioma: a case report.
BMC medical genomicsProgressive encephalomyelitis with rigidity and myoclonus (PERM).
Practical neurologyDelineating the genotypic and phenotypic spectrum of HECW2-related neurodevelopmental disorders.
Journal of medical geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de paraplegia espástica-epilepsia-perturbação do desenvolvimento intelectual.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de paraplegia espástica-epilepsia-perturbação do desenvolvimento intelectual
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Expanding the Phenotypic Spectrum of the Recurrent De Novo FBXO31 p.Asp334Asn Variant: Evidence for a Novel Neurodevelopmental Disorder (Kruer Syndrome).
- Multisystem manifestations of Sjögren-Larsson syndrome in early childhood and its dental implications.
- CTNNB1-related disorders: clinical and radiological contributions from a French cohort.
- Neuroradiological Phenotype Expansion of the Siddiqi Syndrome: A Case Report.
- Bi-allelic variants in FSD1L cause a neurodevelopmental disorder overlapping with L1 syndrome.
- Biallelic TTBK1 variant causes a severe syndromic neurodevelopmental disorder: clinical and genetic insights from two siblings.
- The first description of CTNNB1 syndrome in the Tunisian population: clinical investigation, molecular docking and molecular dynamics simulation of β-catenin/E-cadherin complex.
- Gait abnormalities in children with FOXP1 syndrome: A case series.
- New Insights Into TRMT10A Syndrome: Case Report and Literature Review.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2816(Orphanet)
- MONDO:0008439(MONDO)
- Epilepsia(PCDT · Ministério da Saúde)
- GARD:4915(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q55345676(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
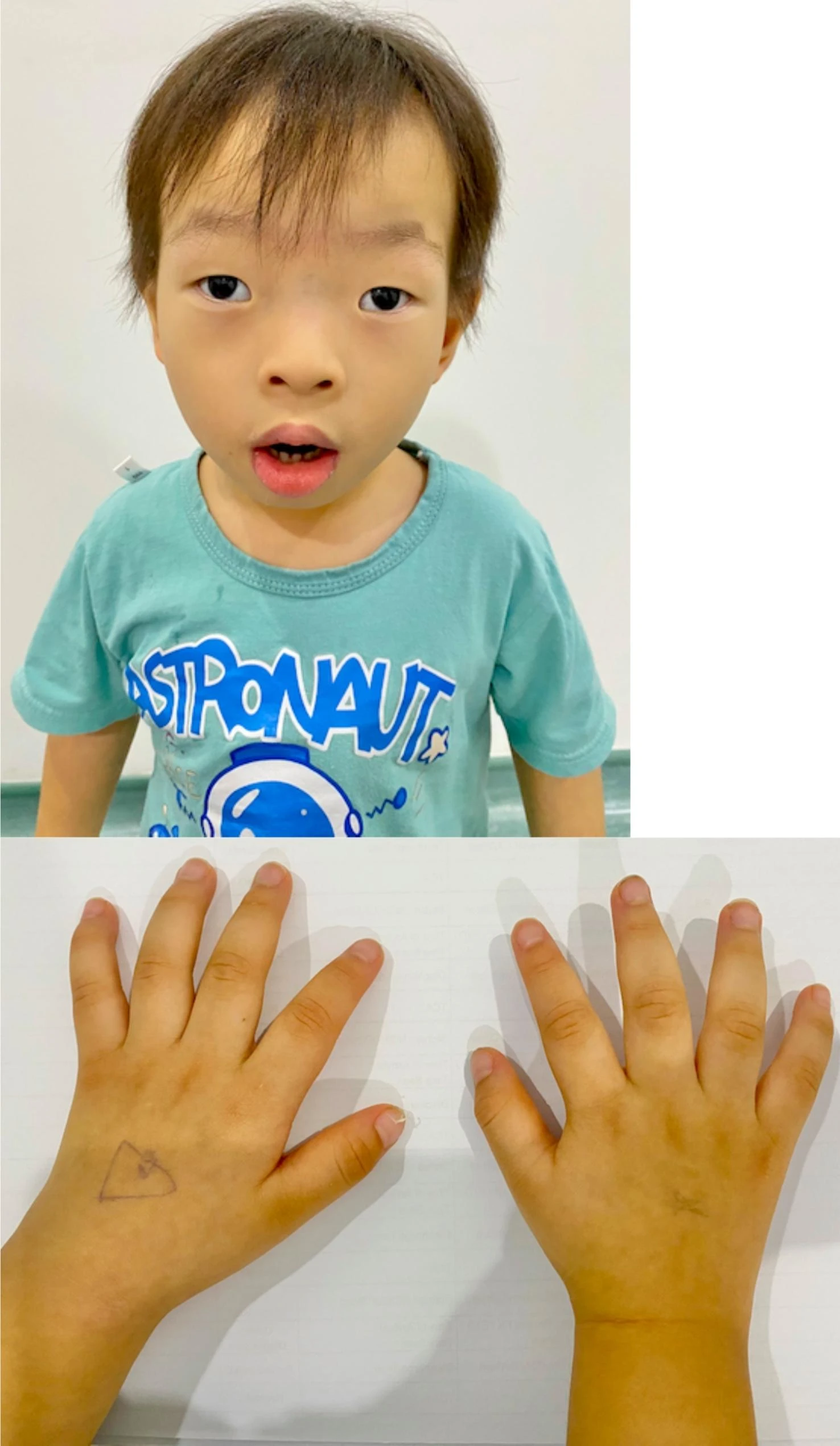
Síndrome de paraplegia espástica-epilepsia-perturbação do desenvolvimento intelectual
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub