Doença rara autossômica dominante, caracterizada por fraqueza e atrofia muscular progressivas, espasmos e contraturas (dedo em martelo, pé plano/cavo), com início na vida adulta. Afeta os neurônios motores inferiores, levando à perda de controle muscular.
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome do neurônio motor inferior com início tardio no adulto é uma doença neuromuscular rara, caracterizada por fraqueza e atrofia muscular progressivas que afetam principalmente os neurônios motores inferiores. A condição tem prevalência estimada em menos de 1 caso por 1.000.000 de pessoas na população geral.[1][4]
A doença geralmente se manifesta na idade adulta e segue um padrão de herança autossômico dominante, o que significa que uma cópia alterada do gene responsável já é suficiente para causar a condição.[1][4]
Sinais e sintomas
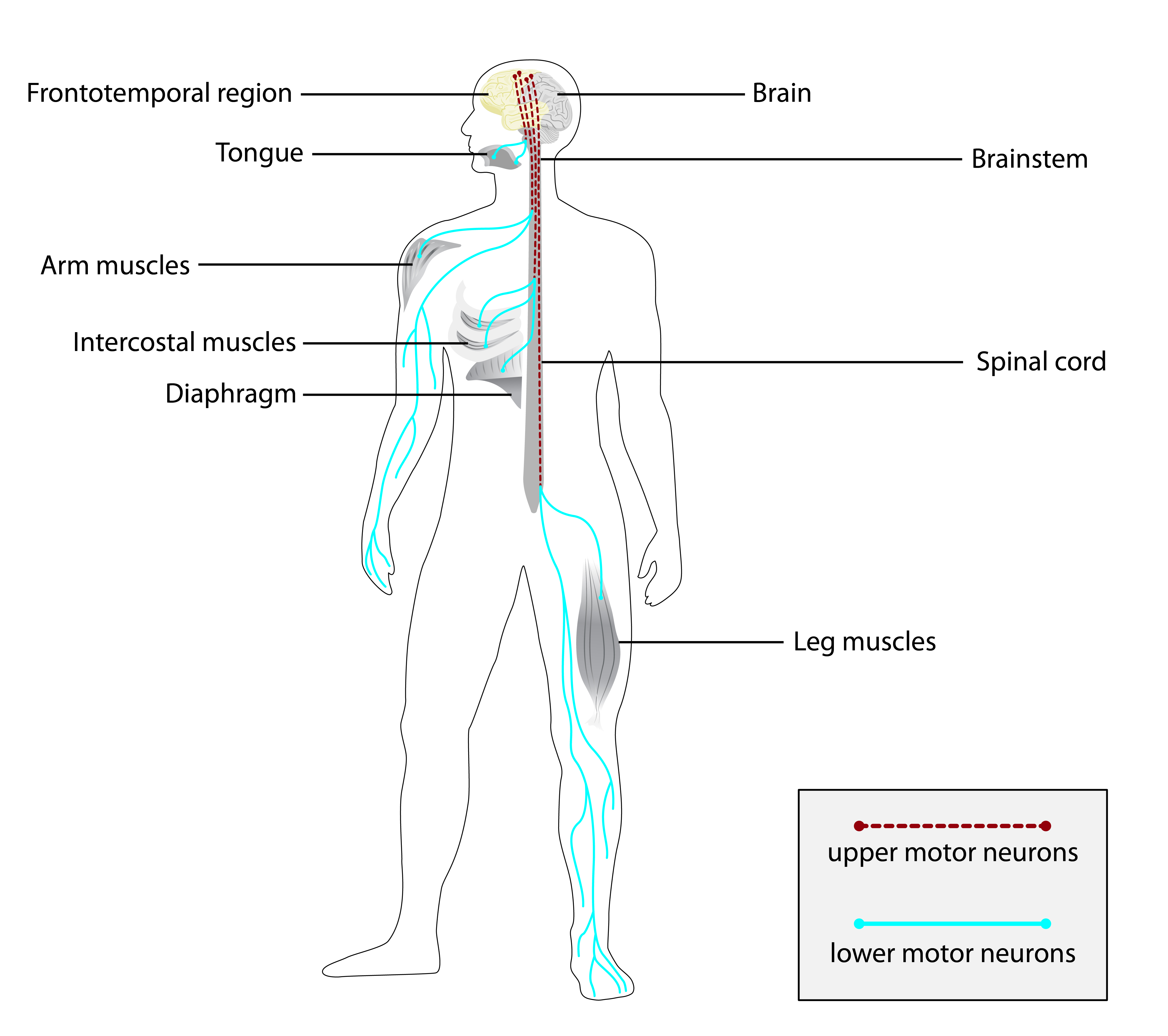
Os sintomas mais comuns incluem fraqueza muscular proximal (mais próxima do tronco) e distal (nas extremidades), tanto nos membros superiores quanto inferiores. Muitas pessoas apresentam fasciculações (contrações musculares involuntárias visíveis sob a pele), cãibras musculares induzidas por exercício ou pelo frio, e arreflexia ou hiporreflexia (reflexos diminuídos ou ausentes).[1][4]
Outros sinais frequentes são atrofia dos músculos intrínsecos da mão, mialgia (dor muscular) no gastrocnêmio (panturrilha), tremor, distúrbio da marcha e incapacidade de andar. Pode haver também disfagia (dificuldade para engolir) e outros sinais bulbares, como fasciculações da língua. Em alguns casos, ocorre comprometimento da sensação vibratória distal e anormalidades na velocidade de condução nervosa sensorial.[1][4]
Exames complementares como a eletroneuromiografia (EMG) podem mostrar tanto alterações neuropáticas quanto miopáticas. A biópsia muscular pode revelar fibras musculares vermelhas rasgadas, vacúolos com bordas e aumento da gordura intramuscular. Em estágios avançados, pode haver anormalidades do sistema respiratório.[1][4]
Causas genéticas
A síndrome é causada por variantes (mutações) no gene CHCHD10, que fornece instruções para a produção da proteína Coiled-coil-helix-coiled-coil-helix domain-containing protein 10, mitocondrial. Essa proteína está localizada na mitocôndria e desempenha um papel importante na função e na sobrevivência dos neurônios motores.[1][2][5]
A herança é autossômica dominante: se um dos pais é portador da variante genética, cada filho tem 50% de chance de herdar a condição. No entanto, a penetrância (probabilidade de desenvolver sintomas) pode ser incompleta, e a expressão da doença pode variar mesmo dentro de uma mesma família.[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sintomas, nos achados de exames como eletroneuromiografia (EMG) e biópsia muscular, e é confirmado pelo teste genético molecular. O sequenciamento completo do exoma (WES) é um dos procedimentos disponíveis para identificar variantes no gene CHCHD10.[1][2][5]
Atualmente, há 16 testes genéticos disponíveis para essa condição e 298 variantes descritas no ClinVar, um banco de dados público que reúne informações sobre variantes genéticas e sua relação com doenças.[1][2][5]
Tratamento e manejo
Não há cura conhecida para a Síndrome do neurônio motor inferior com início tardio no adulto. O tratamento é multidisciplinar e foca no alívio dos sintomas, na manutenção da função muscular e na melhora da qualidade de vida. O acompanhamento com fisioterapia, terapia ocupacional e fonoaudiologia pode ser benéfico para lidar com a fraqueza muscular, a disfagia e as dificuldades de marcha.[1][2]
No âmbito do Sistema Único de Saúde (SUS), o atendimento em reabilitação para doenças raras está disponível. Além disso, medicamentos como nusinersena (Spinraza) e risdiplam (Evrysdi), originalmente desenvolvidos para a atrofia muscular espinhal (AME), podem ser considerados em alguns contextos, conforme avaliação médica especializada.[1][2]
Prognóstico e qualidade de vida
O prognóstico é variável. A progressão da fraqueza muscular pode levar à perda da capacidade de andar e, em alguns casos, ao comprometimento respiratório. O acompanhamento regular com uma equipe multidisciplinar é essencial para monitorar a evolução da doença e ajustar as estratégias de suporte.[1][2]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Doença rara autossômica dominante, caracterizada por fraqueza e atrofia muscular progressivas, espasmos e contraturas (dedo em martelo, pé plano/cavo), com início na vida adulta. Afeta os neurônios motores inferiores, levando à perda de controle muscular.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome do neurônio motor inferior com início tardio no adulto é uma doença neuromuscular rara, caracterizada por fraqueza e atrofia muscular progressivas que afetam principalmente os neurônios motores inferiores. A condição tem prevalência estimada em menos de 1 caso por 1.000.000 de pessoas na população geral.[1][4]
A doença geralmente se manifesta na idade adulta e segue um padrão de herança autossômico dominante, o que significa que uma cópia alterada do gene responsável já é suficiente para causar a condição.[1][4]
Sinais e sintomas
Os sintomas mais comuns incluem fraqueza muscular proximal (mais próxima do tronco) e distal (nas extremidades), tanto nos membros superiores quanto inferiores. Muitas pessoas apresentam fasciculações (contrações musculares involuntárias visíveis sob a pele), cãibras musculares induzidas por exercício ou pelo frio, e arreflexia ou hiporreflexia (reflexos diminuídos ou ausentes).[1][4]
Outros sinais frequentes são atrofia dos músculos intrínsecos da mão, mialgia (dor muscular) no gastrocnêmio (panturrilha), tremor, distúrbio da marcha e incapacidade de andar. Pode haver também disfagia (dificuldade para engolir) e outros sinais bulbares, como fasciculações da língua. Em alguns casos, ocorre comprometimento da sensação vibratória distal e anormalidades na velocidade de condução nervosa sensorial.[1][4]
Exames complementares como a eletroneuromiografia (EMG) podem mostrar tanto alterações neuropáticas quanto miopáticas. A biópsia muscular pode revelar fibras musculares vermelhas rasgadas, vacúolos com bordas e aumento da gordura intramuscular. Em estágios avançados, pode haver anormalidades do sistema respiratório.[1][4]
Causas genéticas
A síndrome é causada por variantes (mutações) no gene CHCHD10, que fornece instruções para a produção da proteína Coiled-coil-helix-coiled-coil-helix domain-containing protein 10, mitocondrial. Essa proteína está localizada na mitocôndria e desempenha um papel importante na função e na sobrevivência dos neurônios motores.[1][2][5]
A herança é autossômica dominante: se um dos pais é portador da variante genética, cada filho tem 50% de chance de herdar a condição. No entanto, a penetrância (probabilidade de desenvolver sintomas) pode ser incompleta, e a expressão da doença pode variar mesmo dentro de uma mesma família.[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sintomas, nos achados de exames como eletroneuromiografia (EMG) e biópsia muscular, e é confirmado pelo teste genético molecular. O sequenciamento completo do exoma (WES) é um dos procedimentos disponíveis para identificar variantes no gene CHCHD10.[1][2][5]
Atualmente, há 16 testes genéticos disponíveis para essa condição e 298 variantes descritas no ClinVar, um banco de dados público que reúne informações sobre variantes genéticas e sua relação com doenças.[1][2][5]
Tratamento e manejo
Não há cura conhecida para a Síndrome do neurônio motor inferior com início tardio no adulto. O tratamento é multidisciplinar e foca no alívio dos sintomas, na manutenção da função muscular e na melhora da qualidade de vida. O acompanhamento com fisioterapia, terapia ocupacional e fonoaudiologia pode ser benéfico para lidar com a fraqueza muscular, a disfagia e as dificuldades de marcha.[1][2]
No âmbito do Sistema Único de Saúde (SUS), o atendimento em reabilitação para doenças raras está disponível. Além disso, medicamentos como nusinersena (Spinraza) e risdiplam (Evrysdi), originalmente desenvolvidos para a atrofia muscular espinhal (AME), podem ser considerados em alguns contextos, conforme avaliação médica especializada.[1][2]
Prognóstico e qualidade de vida
O prognóstico é variável. A progressão da fraqueza muscular pode levar à perda da capacidade de andar e, em alguns casos, ao comprometimento respiratório. O acompanhamento regular com uma equipe multidisciplinar é essencial para monitorar a evolução da doença e ajustar as estratégias de suporte.[1][2]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 16 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 39 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisMay be involved in the maintenance of mitochondrial organization and mitochondrial cristae structure
Mitochondrion intermembrane space
Frontotemporal dementia and/or amyotrophic lateral sclerosis 2
A neurodegenerative disorder characterized by frontotemporal dementia and/or amyotrophic lateral sclerosis in affected individuals. There is high intrafamilial variation. Frontotemporal dementia is characterized by frontal and temporal lobe atrophy associated with neuronal loss, gliosis, and dementia. Patients exhibit progressive changes in social, behavioral, and/or language function. Amyotrophic lateral sclerosis is characterized by the death of motor neurons in the brain, brainstem, and spinal cord, resulting in fatal paralysis.
Variantes genéticas (ClinVar)
298 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 222 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome do neurônio motor inferior com início tardio no adulto
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome do neurônio motor inferior com início tardio no adulto.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome do neurônio motor inferior com início tardio no adulto
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:276435(Orphanet)
- OMIM OMIM:615048(OMIM)
- MONDO:0014025(MONDO)
- GARD:17282(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Q25451213(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome do neurônio motor inferior com início tardio no adulto
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata