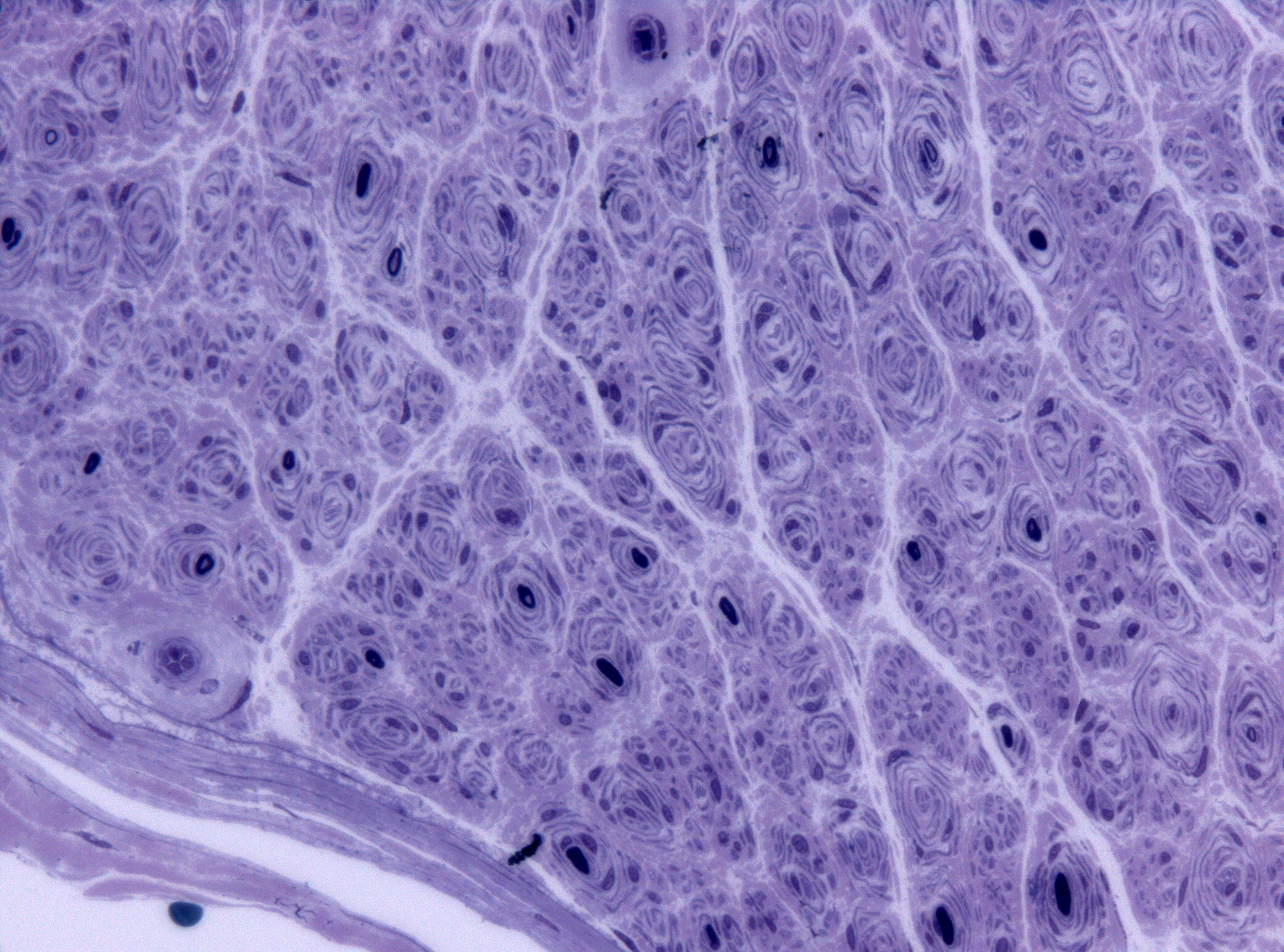
A neuromiotonia, também conhecida como síndrome de Isaacs, é uma síndrome de hiperexcitabilidade dos nervos periféricos (HPN) que se apresenta como atividade motora contínua. Os achados clínicos incluem cãibras, fasciculações e mioquimia. O eletrodiagnóstico desempenha um papel fundamental no diagnóstico, demonstrando pós-descargas nos estudos de condução nervosa e potenciais de fasciculação, descargas mioquímicas, descargas neuromiotónicas e outros tipos de atividade espontânea anormal no exame com agulha. A etiopatogenia envolve a interação de factores genéticos, auto-imunes e paraneoplásicos, o que exige uma avaliação abrangente das causas subjacentes. O tratamento inicial é sintomático, mas a imunoterapia é frequentemente necessária e pode ser eficaz.
Introdução
O que você precisa saber de cara
Doença genética rara que afeta nervos periféricos, causando fraqueza muscular progressiva e perda de sensibilidade. A desmielinização prejudica a transmissão de sinais nervosos, impactando movimentos e sensações.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Neuropatia sensitiva e motora desmielinizante hereditária autossômica dominante
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
A novel KIDINS220 mutation associated with hereditary spastic paraplegia accompanied by severe peripheral neuropathy.
Mutations in KIDINS220 are known to cause hereditary spastic paraplegia (HSP) and SINO syndrome. However, the phenotypic and genotypic spectrum of KIDINS220-related disorders remains incompletely understood. Herein, we describe the clinical, electrophysiological, histopathological, and genetic features of a novel KIDINS220 sterile alpha motif (SAM) -like domain mutation identified in a Chinese family with HSP accompanied by severe peripheral neuropathy (PN). Clinical data, electrophysiological characteristics, and sural nerve histopathology were analyzed in a 19-year-old Chinese male. Genetic testing was performed in his family by using whole-exome sequencing, mitochondrial genome testing, and Sanger validation. A comprehensive literature review was conducted to analyze the phenotypic and genetic data of previously reported cases with KIDINS220 variants up to July 2025. The proband exhibited classical signs of autosomal dominant HSP accompanied by severe multifocal sensory-motor PN. The spinal cord MRI showed mild spinal cord thinning, while the brain MRI and nerve ultrasound examinations were normal. Electrophysiological study revealed absent sensory nerve responses and globally reduced motor conduction velocities. Sural nerve biopsy confirmed significantly reduced nerve fiber density, myelin defects, axonal degeneration, and mitochondrial abnormalities. A heterozygous KIDINS220 c.3668A > G (p. Glu1223Gly) mutation, located within the SAM domain, was identified in both the proband and his mother. A total of 42 cases from 11 cohorts were reviewed. We suggest that patients with KIDINS220 SAM domain mutation may present with HSP accompanied by severe, mixed axonal and demyelinating PN, expanding the existing spectrum of the clinical phenotypes and pathogenic variants of KIDINS220.
A Novel GBF1 Variant in a Charcot-Marie-Tooth Type 2: Insights from Familial Analysis.
Axonal Charcot-Marie-Tooth disease type 2 (CMT2) accounts for 24% of Hereditary Motor/Sensory Peripheral Neuropathies. CMT2 type GG, due to four distinct heterozygous mutations in the Golgi brefeldin A resistant guanine nucleotide exchange factor 1 (GBF1) gene (OMIM 606483), was described in seven cases from four unrelated families with autosomal dominant inheritance. It is characterized by slowly progressive distal muscle weakness and atrophy, primarily affecting the lower limbs. Here, we present two siblings sharing a novel GBF1 variant. Patient II.1 (male, 61 years at onset) presented lower limb hypoesthesia and walking difficulty; the examination revealed a postural tremor, a positive Romberg test, and muscle atrophy in the lower limbs and hands. Patient II.2 (his sister, 59 years at onset) had lower limb dysesthesias, hand paresthesia, and lower-limb stiffness. They underwent clinical evaluations, blood tests, and electroneurography. Their father represents a potentially affected individual, although a genetic analysis was not conducted. All tests for peripheral neuropathies were unremarkable, including metabolic and autoimmune screening. Both showed a mixed demyelinating-axonal sensory-motor neuropathy. Genetic analysis revealed a new heterozygous GBF1 variant of uncertain significance. Based on autosomal dominant inheritance, as well as clinical and physiological features, a possible novel CMT2GG was diagnosed. Further research, including functional assays and in vitro studies, is necessary to confirm this variant's causal link.
Clinical and molecular genetic characteristics of 24 families of hereditary neuropathy with liability to pressure palsy and literature review.
Hereditary neuropathy with liability to pressure palsy (HNPP) is a rare autosomal dominant peripheral neuropathy, usually caused by heterozygous deletion mutations in the peripheral myelin protein 22 (PMP22) gene. This study aims to investigate the clinical and molecular genetic characteristics of HNPP. HNPP patients in the Department of Neurology at Third Xiangya Hospital of Central South University from 2009 to 2023 were included in this study. The general clinical data, nervous electrophysiological and molecular genetic examination results were collected and analyzed. Molecular genetic examination was to screen for deletion of PMP22 gene using multiplex ligation-dependent probe amplification (MLPA) after extracting genomic DNA from peripheral blood; and if no PMP22 deletion mutation was detected, next-generation sequencing was used to screen for PMP22 point mutations. The related literatures of HNPP were reviewed, and the clinical and molecular genetic characteristics of HNPP patients were analyzed. A total of 34 HNPP patients from 24 unrelated Chinese Han families were included in this study, including 25 males and 9 females. The average age at illness onset was 22.0 years. Sixty-two point five percent of the families had a positive family history. Among them, 30 patients had symptoms of peripheral nerve paralysis. Patients often presented with paroxysmal single limb weakness with (or) numbness (25/30), and some patients had paroxysmal unilateral recurrent laryngeal nerve (vagus nerve) paralysis (2/30). Physical examination revealed muscle weakness (23/29), hypoesthesia (9/29), weakened or absent ankle reflexes (20/29), distal limb muscle atrophy (8/29) and high arched feet (5/29). Most patients (26/30) could fully recover to normal after an acute attack. Thirty-one patients in our group underwent nervous electrophysiological examination, and showed multiple demyelinating peripheral neuropathies with both motor and sensory nerves involved. Most patients showed significantly prolonged distal motor latency (DML), mild to moderate nerve conduction velocity slowing, decreased amplitude of compound muscle action potential (CMAP) and sensory nerve action potential (SNAP), and sometimes with conduction block. Nerve motor conduction velocity was (48.5±5.5) m/s, and the CMAP amplitude was (8.4±5.1) mV. Nerve sensory conduction velocity was (37.4±10.5) m/s, and the SNAP amplitude was (14.4±15.2) μV. There were 24 families, 23 of whom had the classical PMP22 deletion, the last one had a heterozygous pathogenic variant in the PMP22 gene sequence (c.434delT). By reviewing clinical data and genetic testing results of reported 1 734 HNPP families, we found that heterozygous deletion mutation of PMP22 was the most common pathogenic mutation of HNPP (93.4%). Other patients were caused by PMP22 small mutations (4.0%), PMP22 heterozygous gross deletions (0.6%), and PMP22 complex rearrangements (0.1%). Thirty-eight sorts of HNPP-related PMP22 small mutations was reported, including missense mutations (10/38), nonsense mutations (4/38), base deletion mutations (13/38), base insertion mutations (3/38), and shear site mutations (8/38). HNPP patients most often presented with episodic painless single nerve palsy. Common peroneal nerve, ulnar nerve, and brachial plexus nerve were the most common involved nerves, accounting for about 75%. Only eighteen patients with cranial nerve involved was reported. Heterozygous deletion mutation of PMP22 is the most common pathogenic mutation of HNPP. Patients is characterized by episodic and painless peripheral nerve paralysis, mainly involving common peroneal nerve, ulnar nerve, and other peripheral nerves. Nervous electrophysiological examination has high sensitivity and specificity for the diagnosis of HNPP, which is manifested by extensive demyelinating changes. For patients with suspected HNPP, nervous electrophysiological examination and PMP22-MLPA detection are preferred. Sanger sequencing or next generation sequencing can be considered to detect other mutations of PMP22. 目的: 遗传性压力易感性周围神经病(hereditary neuropathy with liability to pressure palsy,HNPP)是一种少见的常染色体显性遗传周围神经病,通常由周围髓鞘蛋白22(peripheral myelin protein 22,PMP22)基因杂合缺失突变引起。本研究旨在探讨HNPP患者的临床和分子遗传学特征。方法: 纳入2009至2023年就诊于中南大学湘雅三医院神经内科的HNPP患者,收集并分析患者的一般临床资料、神经电生理和分子遗传学检查结果。分子遗传学检查为提取外周血基因组DNA后采用多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)进行PMP22大片段缺失的筛查;若未检测到PMP22缺失突变,则用二代测序法筛查PMP22点突变。进一步对HNPP相关文献进行回顾,分析HNPP患者的临床和分子遗传学特征。结果: 共纳入来自24个无血缘关系的中国汉族家系的34例HNPP患者,包括25名男性和9名女性,平均22.0岁起病,有阳性家族史的家系占62.5%。30例患者出现周围神经麻痹的症状,常表现为发作性单肢无力伴/或麻木(25/30),亦可表现为发作性单侧喉返神经(迷走神经)麻痹(2/30)。体格检查可发现受累神经相应支配区域的肌肉无力(23/29)和浅感觉减退(9/29),踝反射减弱或消失(20/29),肢体远端肌肉萎缩(8/29)及高弓足(5/29)。多数患者(26/30)在急性发作后可完全恢复正常。有31例患者完成神经电生理检查,均表现为多发性周围神经损害,以运动及感觉神经髓鞘损害为主,多数患者可有远端运动潜伏期(distal motor latency,DML)明显延长,轻至中度的神经传导速度减慢,复合肌肉动作电位(compound muscle action potential,CMAP)及感觉神经动作电位(sensory nerve action potential,SNAP)波幅降低,有时可出现传导阻滞。正中神经运动传导速度为(48.5±5.5) m/s,CMAP波幅为(8.4±5.1) mV;正中神经感觉传导速度为(37.4±10.5) m/s,SNAP波幅为(14.4±13.0) μV。在24个HNPP家系中,经MLPA检测证实有23个家系为PMP22杂合缺失突变导致,经二代测序证实剩余1个家系为PMP22 c.434delT突变所致。文献检索到1 734个进行了基因检测的HNPP确诊家系,其基因检测结果再次证实了PMP22杂合缺失突变是最常见的突变类型,占93.4%,其余突变类型包括PMP22微小突变(4.0%)、PMP22杂合不完全缺失突变(0.6%)、PMP22复杂易位突变(0.1%)。目前HNPP相关的PMP22微小突变共报道38种,包括错义突变(10/38)、无义突变(4/38)、碱基缺失突变(13/38)、碱基插入突变(3/38)、剪切位点突变(8/38)。HNPP患者最常表现为发作性无痛性单神经麻痹,腓总神经、尺神经和臂丛神经受累最为常见,约占75%。颅神经受累的患者仅有18例报道。结论: PMP22杂合缺失突变是HNPP最常见的突变类型。HNPP以发作性无痛性单神经麻痹为主要特点,主要累及腓总神经、尺神经等周围神经。神经电生理检查对于诊断本病具有较高的灵敏度和特异度,表现为广泛的脱髓鞘改变。对于怀疑HNPP的患者,首选完善神经电生理检查及PMP22大片段杂合缺失的检测。必要时可以完善Sanger测序或二代测序,对PMP22其他突变类型进行检测以防漏诊。. Hereditary neuropathy with liability to pressure palsy (HNPP) is a rare autosomal dominant peripheral neuropathy, usually caused by heterozygous deletion mutations in the peripheral myelin protein 22 (PMP22) gene. This study aims to investigate the clinical and molecular genetic characteristics of HNPP. HNPP patients in the Department of Neurology at Third Xiangya Hospital of Central South University from 2009 to 2023 were included in this study. The general clinical data, nervous electrophysiological and molecular genetic examination results were collected and analyzed. Molecular genetic examination was to screen for deletion of PMP22 gene using multiplex ligation-dependent probe amplification (MLPA) after extracting genomic DNA from peripheral blood; and if no PMP22 deletion mutation was detected, next-generation sequencing was used to screen for PMP22 point mutations. The related literatures of HNPP were reviewed, and the clinical and molecular genetic characteristics of HNPP patients were analyzed. A total of 34 HNPP patients from 24 unrelated Chinese Han families were included in this study, including 25 males and 9 females. The average age at illness onset was 22.0 years. Sixty-two point five percent of the families had a positive family history. Among them, 30 patients had symptoms of peripheral nerve paralysis. Patients often presented with paroxysmal single limb weakness with (or) numbness (25/30), and some patients had paroxysmal unilateral recurrent laryngeal nerve (vagus nerve) paralysis (2/30). Physical examination revealed muscle weakness (23/29), hypoesthesia (9/29), weakened or absent ankle reflexes (20/29), distal limb muscle atrophy (8/29) and high arched feet (5/29). Most patients (26/30) could fully recover to normal after an acute attack. Thirty-one patients in our group underwent nervous electrophysiological examination, and showed multiple demyelinating peripheral neuropathies with both motor and sensory nerves involved. Most patients showed significantly prolonged distal motor latency (DML), mild to moderate nerve conduction velocity slowing, decreased amplitude of compound muscle action potential (CMAP) and sensory nerve action potential (SNAP), and sometimes with conduction block. Nerve motor conduction velocity was (48.5±5.5) m/s, and the CMAP amplitude was (8.4±5.1) mV. Nerve sensory conduction velocity was (37.4±10.5) m/s, and the SNAP amplitude was (14.4±15.2) μV. There were 24 families, 23 of whom had the classical PMP22 deletion, the last one had a heterozygous pathogenic variant in the PMP22 gene sequence (c.434delT). By reviewing clinical data and genetic testing results of reported 1 734 HNPP families, we found that heterozygous deletion mutation of PMP22 was the most common pathogenic mutation of HNPP (93.4%). Other patients were caused by PMP22 small mutations (4.0%), PMP22 heterozygous gross deletions (0.6%), and PMP22 complex rearrangements (0.1%). Thirty-eight sorts of HNPP-related PMP22 small mutations was reported, including missense mutations (10/38), nonsense mutations (4/38), base deletion mutations (13/38), base insertion mutations (3/38), and shear site mutations (8/38). HNPP patients most often presented with episodic painless single nerve palsy. Common peroneal nerve, ulnar nerve, and brachial plexus nerve were the most common involved nerves, accounting for about 75%. Only eighteen patients with cranial nerve involved was reported. Heterozygous deletion mutation of PMP22 is the most common pathogenic mutation of HNPP. Patients is characterized by episodic and painless peripheral nerve paralysis, mainly involving common peroneal nerve, ulnar nerve, and other peripheral nerves. Nervous electrophysiological examination has high sensitivity and specificity for the diagnosis of HNPP, which is manifested by extensive demyelinating changes. For patients with suspected HNPP, nervous electrophysiological examination and PMP22-MLPA detection are preferred. Sanger sequencing or next generation sequencing can be considered to detect other mutations of PMP22.
Multiple tendon transfer for a case of radial nerve palsy in hereditary neuropathy with liability to pressure palsy.
Hereditary neuropathy with liability to pressure palsy (HNPP) is a rare autosomal dominant disease characterized by focal, recurrent, demyelinating peripheral neuropathies. It is caused by deletions of the gene encoding for peripheral myelin protein 22 (PMP22) on chromosome 17. While it may range widely, the most common clinical presentation is an acute, focal mononeuropathy with numbness or muscle weakness after trauma or compression. Diagnostic tools include electrophysiological studies, genetic tests and nerve biopsies. There is no standard surgical or pharmacological treatment. The course of the disease is usually benign, with spontaneous improvement after most episodes of peripheral nerve palsy. HNPP is best managed by early detection, preventative measures, and subsequent treatment of symptoms. According to the medical literature, operative treatment was undertaken in few cases and limited to decompression of the nerve at the classic entrapment sites of the carpal or cubital tunnels. We present a case of multiple tendon transfer (pronator teres to extensor carpi radialis brevis and flexor carpi radialis to extensor digitorum communis) with a two-year follow-up in a 24-year-old woman with HNPP who was affected by irreversible radial nerve palsy, and conclude with a review of the medical literature related to the disease.
Differential effects of Mendelian GDAP1 clinical variants on mitochondria-lysosome membrane contacts sites.
GDAP1 pathogenic variants cause Charcot-Marie-Tooth (CMT) disease, the most common hereditary motor and sensory neuropathy. CMT-GDAP1 can be axonal or demyelinating, with autosomal dominant or recessive inheritance, leading to phenotypic heterogeneity. Recessive GDAP1 variants cause a severe phenotype, whereas dominant variants are associated with a milder disease course. GDAP1 is an outer mitochondrial membrane protein involved in mitochondrial membrane contact sites (MCSs) with the plasmatic membrane, the endoplasmic reticulum (ER), and lysosomes. In GDAP1-deficient models, the pathophysiology includes morphological defects in mitochondrial network and ER, impaired Ca2+ homeostasis, oxidative stress, and mitochondrial MCSs defects. Nevertheless, the underlying pathophysiology of dominant variants is less understood. Here, we study the effect upon mitochondria-lysosome MCSs of two GDAP1 clinical variants located in the α-loop interaction domain of the protein. p.Thr157Pro dominant variant causes the increase in these MCSs that correlates with a hyper-fissioned mitochondrial network. In contrast, p.Arg161His recessive variant, which is predicted to significantly change the contact surface of GDAP1, causes decreased contacts with more elongated mitochondria. Given that mitochondria-lysosome MCSs regulate Ca2+ transfer from the lysosome to mitochondria, our results support that GDAP1 clinical variants have different consequences for Ca2+ handling and that could be primary insults determining differences in severity between dominant and recessive forms of the disease.
📚 EuropePMCmostrando 21
A novel KIDINS220 mutation associated with hereditary spastic paraplegia accompanied by severe peripheral neuropathy.
Frontiers in neuroscienceA Novel GBF1 Variant in a Charcot-Marie-Tooth Type 2: Insights from Familial Analysis.
GenesClinical and molecular genetic characteristics of 24 families of hereditary neuropathy with liability to pressure palsy and literature review.
Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciencesMultiple tendon transfer for a case of radial nerve palsy in hereditary neuropathy with liability to pressure palsy.
Nagoya journal of medical scienceDifferential effects of Mendelian GDAP1 clinical variants on mitochondria-lysosome membrane contacts sites.
Biology openHereditary neuropathy with liability to pressure palsies (HNPP): Intrafamilial phenotypic variability and early childhood refusal to walk as the presenting symptom.
Italian journal of pediatricsThe Antiepileptic Valproic Acid Ameliorates Charcot-Marie-Tooth 2W (CMT2W) Disease-Associated HARS1 Mutation-Induced Inhibition of Neuronal Cell Morphological Differentiation Through c-Jun N-terminal Kinase.
Neurochemical researchWhole-exome sequencing identifies a heterozygous mutation in SLC12A6 associated with hereditary sensory and motor neuropathy.
Neuromuscular disorders : NMDDemyelination in hereditary sensory neuropathy type-1C.
Annals of clinical and translational neurologyHereditary sensory and autonomic neuropathy type IC accompanied by upper motor neuron abnormalities and type II juxtafoveal retinal telangiectasias.
Journal of the peripheral nervous system : JPNSPeripheral neuropathy in diabetes: it's not always what it looks like.
Diabetic medicine : a journal of the British Diabetic AssociationTransthyretin familial amyloid polyneuropathy (TTR-FAP): Parameters for early diagnosis.
Brain and behaviorClinical, electrophysiological, genetic, and imaging features of six Chinese Han patients with hereditary neuropathy with liability to pressure palsies (HNPP).
Journal of clinical neuroscience : official journal of the Neurosurgical Society of AustralasiaHereditary neuropathy with liability to pressure palsy (HNPP): report of a family with a new point mutation in PMP22 gene.
Italian journal of pediatricsA novel missense variant (Gln220Arg) of GNB4 encoding guanine nucleotide-binding protein, subunit beta-4 in a Japanese family with autosomal dominant motor and sensory neuropathy.
European journal of medical geneticsUnilateral oculomotor palsy in Charcot-Marie-Tooth disease 1A (CMT 1A).
Clinical neurology and neurosurgeryPhenotypic spectrum of Charcot-Marie-Tooth disease due to LITAF/SIMPLE mutations: a study of 18 patients.
European journal of neurology[Hereditary neuropathy with liability to pressure palsies in childhood: Report of three cases].
Archives de pediatrie : organe officiel de la Societe francaise de pediatrie[Phenotypes of Charcot-Marie-Tooth Syndrome and Differential Diagnosis Focused in Inflammatory Neuropathies].
Brain and nerve = Shinkei kenkyu no shinpoLoss of function mutations in HARS cause a spectrum of inherited peripheral neuropathies.
Brain : a journal of neurologyMitochondrial dynamics and inherited peripheral nerve diseases.
Neuroscience lettersAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Neuropatia sensitiva e motora desmielinizante hereditária autossômica dominante.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Neuropatia sensitiva e motora desmielinizante hereditária autossômica dominante
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A novel KIDINS220 mutation associated with hereditary spastic paraplegia accompanied by severe peripheral neuropathy.
- A Novel GBF1 Variant in a Charcot-Marie-Tooth Type 2: Insights from Familial Analysis.
- Clinical and molecular genetic characteristics of 24 families of hereditary neuropathy with liability to pressure palsy and literature review.Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciences· 2023· PMID 38432886mais citado
- Multiple tendon transfer for a case of radial nerve palsy in hereditary neuropathy with liability to pressure palsy.
- Differential effects of Mendelian GDAP1 clinical variants on mitochondria-lysosome membrane contacts sites.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:140453(Orphanet)
- MONDO:0015359(MONDO)
- Q55785419(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar