A Displasia Arritmogênica do Ventrículo Direito (DAVD) Familiar é o tipo de DAVD de origem genética, passado de pais para filhos com um padrão de herança autossômico dominante. É uma doença do músculo do coração marcada por arritmias ventriculares (batimentos cardíacos irregulares e perigosos que vêm das câmaras inferiores do coração) que podem ser fatais, apresentando um padrão elétrico específico chamado bloqueio de ramo esquerdo. Os sintomas podem incluir palpitações, taquicardia ventricular (batimentos muito acelerados), desmaios e até morte súbita. A doença é causada pela degeneração e substituição do músculo do ventrículo direito por tecido gorduroso e fibroso, o que pode resultar na formação de dilatações ou "sacos" na parede desse ventrículo (aneurismas).
Introdução
O que você precisa saber de cara
A Displasia Arritmogênica do Ventrículo Direito (DAVD) Familiar é o tipo de DAVD de origem genética, passado de pais para filhos com um padrão de herança autossômico dominante. É uma doença do músculo do coração marcada por arritmias ventriculares (batimentos cardíacos irregulares e perigosos que vêm das câmaras inferiores do coração) que podem ser fatais, apresentando um padrão elétrico específico chamado bloqueio de ramo esquerdo. Os sintomas podem incluir palpitações, taquicardia ventricular (batimentos muito acelerados), desmaios e até morte súbita. A doença é causada pela degeneração e substituição do músculo do ventrículo direito por tecido gorduroso e fibroso, o que pode resultar na formação de dilatações ou "sacos" na parede desse ventrículo (aneurismas).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 18 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 45 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
14 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisReversibly inhibits the activity of ATP2A2/SERCA2 in cardiac sarcoplasmic reticulum by decreasing the apparent affinity of the ATPase for Ca(2+) (PubMed:28890335). Binds preferentially to the ATP-bound E1 conformational form of ATP2A2 which predominates at low Ca(2+) concentrations during the diastolic phase of the cardiac cycle (By similarity). Inhibits ATP2A2 Ca(2+) affinity by disrupting its allosteric activation by ATP (By similarity). Modulates the contractility of the heart muscle in respo
Endoplasmic reticulum membraneSarcoplasmic reticulum membraneMitochondrion membraneMembrane
Cardiomyopathy, dilated, 1P
A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death.
May function as an adapter in striated muscle to couple protein kinase C-mediated signaling via its LIM domains to the cytoskeleton
Cytoplasm, perinuclear regionCell projection, pseudopodiumCytoplasm, cytoskeletonCytoplasm, myofibril, sarcomere, Z line
Cardiomyopathy, dilated, 1C, with or without left ventricular non-compaction
A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. Cardiomyopathy dilated type 1C is associated with left ventricular non-compaction in some patients. Left ventricular non-compaction is characterized by numerous prominent trabeculations and deep intertrabecular recesses in hypertrophied and hypokinetic segments of the left ventricle.
Cytosolic calcium-activated calcium channel that mediates the release of Ca(2+) from the sarcoplasmic reticulum into the cytosol and thereby plays a key role in triggering cardiac muscle contraction. Aberrant channel activation can lead to cardiac arrhythmia. In cardiac myocytes, calcium release is triggered by increased Ca(2+) cytosolic levels due to activation of the L-type calcium channel CACNA1C. The calcium channel activity is modulated by formation of heterotetramers with RYR3. Required fo
Sarcoplasmic reticulum membrane
Ventricular tachycardia, catecholaminergic polymorphic, 1, with or without atrial dysfunction and/or dilated cardiomyopathy
An arrhythmogenic disorder characterized by stress-induced, bidirectional ventricular tachycardia that may degenerate into cardiac arrest and cause sudden death. Patients present with recurrent syncope, seizures, or sudden death after physical activity or emotional stress. CPVT1 inheritance is autosomal dominant.
Calcium-dependent cell adhesion protein; preferentially mediates homotypic cell-cell adhesion by dimerization with a CDH2 chain from another cell. Cadherins may thus contribute to the sorting of heterogeneous cell types. Acts as a regulator of neural stem cells quiescence by mediating anchorage of neural stem cells to ependymocytes in the adult subependymal zone: upon cleavage by MMP24, CDH2-mediated anchorage is affected, leading to modulate neural stem cell quiescence. Plays a role in cell-to-
Cell membraneCell membrane, sarcolemmaCell junctionCell surfaceCell junction, desmosomeCell junction, adherens junctionCell projection, axon
Arrhythmogenic right ventricular dysplasia, familial, 14
A congenital heart disease characterized by infiltration of adipose and fibrous tissue into the right ventricle and loss of myocardial cells, resulting in ventricular and supraventricular arrhythmias.
A component of desmosome cell-cell junctions which are required for positive regulation of cellular adhesion (PubMed:17559062, PubMed:38395410). Involved in the interaction of plaque proteins and intermediate filaments mediating cell-cell adhesion. Required for proliferation and viability of embryonic stem cells in the blastocyst, thereby crucial for progression of post-implantation embryonic development (By similarity). Maintains pluripotency by regulating epithelial to mesenchymal transition/m
Cell membraneCell junction, desmosomeCytoplasm
Arrhythmogenic right ventricular dysplasia, familial, 10
A congenital heart disease characterized by infiltration of adipose and fibrous tissue into the right ventricle and loss of myocardial cells, resulting in ventricular and supraventricular arrhythmias.
May be involved in formation of stretch-resistant cell-cell adhesion complexes
Cytoplasm, cytoskeletonCell junction, desmosome
Arrhythmogenic right ventricular dysplasia, familial, 13
A congenital heart disease characterized by infiltration of adipose and fibrous tissue into the right ventricle and loss of myocardial cells, resulting in ventricular and supraventricular arrhythmias.
Lamins are intermediate filament proteins that assemble into a filamentous meshwork, and which constitute the major components of the nuclear lamina, a fibrous layer on the nucleoplasmic side of the inner nuclear membrane (PubMed:10080180, PubMed:10580070, PubMed:10587585, PubMed:10814726, PubMed:11799477, PubMed:12075506, PubMed:12927431, PubMed:15317753, PubMed:18551513, PubMed:18611980, PubMed:2188730, PubMed:22431096, PubMed:2344612, PubMed:23666920, PubMed:24741066, PubMed:31434876, PubMed:
Nucleus laminaNucleus envelopeNucleus, nucleoplasmNucleus matrixNucleus speckle
Emery-Dreifuss muscular dystrophy 2, autosomal dominant
A form of Emery-Dreifuss muscular dystrophy, a degenerative myopathy characterized by weakness and atrophy of muscle without involvement of the nervous system, early contractures of the elbows, Achilles tendons and spine, and cardiomyopathy associated with cardiac conduction defects.
A component of desmosome cell-cell junctions which are required for positive regulation of cellular adhesion (PubMed:33596089). Promotes timely incorporation of DSG2 into desmosome intercellular junctions and promotes interaction of desmosome cell junctions with intermediate filament cytokeratin, via modulation of DSP phosphorylation (PubMed:33596089). Plays an important role in desmosome-mediated maintenance of intestinal epithelial cell intercellular adhesion strength and barrier function (Pub
Cell membraneCell junction, desmosome
Arrhythmogenic right ventricular dysplasia, familial, 11
A congenital heart disease characterized by infiltration of adipose and fibrous tissue into the right ventricle and loss of myocardial cells, resulting in ventricular and supraventricular arrhythmias.
May have an important role in maintaining nuclear envelope structure by organizing protein complexes at the inner nuclear membrane. Required for retaining emerin at the inner nuclear membrane (By similarity). Plays a role in the modulation of innate immune signaling through the cGAS-STING pathway by interacting with RNF26 (PubMed:32614325). In addition, functions as a critical signaling component in mediating NF-kappa-B activation by acting downstream of EGFR and upstream of CARD10 (PubMed:27991
Endoplasmic reticulum membraneNucleus inner membraneCell membrane
Arrhythmogenic right ventricular dysplasia, familial, 5
A congenital heart disease characterized by infiltration of adipose and fibrous tissue into the right ventricle and loss of myocardial cells, resulting in ventricular and supraventricular arrhythmias.
Transforming growth factor beta-3 proprotein: Precursor of the Latency-associated peptide (LAP) and Transforming growth factor beta-3 (TGF-beta-3) chains, which constitute the regulatory and active subunit of TGF-beta-3, respectively Required to maintain the Transforming growth factor beta-3 (TGF-beta-3) chain in a latent state during storage in extracellular matrix (By similarity). Associates non-covalently with TGF-beta-3 and regulates its activation via interaction with 'milieu molecules', su
Secreted, extracellular space, extracellular matrixSecreted
Arrhythmogenic right ventricular dysplasia, familial, 1
A congenital heart disease characterized by infiltration of adipose and fibrous tissue into the right ventricle and loss of myocardial cells, resulting in ventricular and supraventricular arrhythmias.
A component of desmosome cell-cell junctions which are required for positive regulation of cellular adhesion (PubMed:25733715). Critical for cell-cell adhesion in early stage blastocysts and progression through proamniotic cavity formation (By similarity). Not required for preimplantation morphogenic process in blastocysts (By similarity). Required for keratin filament anchoring at the desmosome junction and subsequent organization of the keratin intermediate filament network within the cytoplas
Cell projection, axonCell junction, desmosomeCell membraneCytoplasmNucleus
Keratoderma, palmoplantar, striate 2
A dermatological disorder characterized by thickening of the skin on the palms (linear pattern) and the soles (island-like pattern) and flexor aspect of the fingers. Abnormalities of the nails, the teeth and the hair are rarely present.
Key component in the assembly and functioning of vertebrate striated muscles. By providing connections at the level of individual microfilaments, it contributes to the fine balance of forces between the two halves of the sarcomere. The size and extensibility of the cross-links are the main determinants of sarcomere extensibility properties of muscle. In non-muscle cells, seems to play a role in chromosome condensation and chromosome segregation during mitosis. Might link the lamina network to ch
CytoplasmNucleus
Myopathy, myofibrillar, 9, with early respiratory failure
An autosomal dominant myopathy characterized by adulthood onset of weakness in proximal, distal, axial and respiratory muscles. Pelvic girdle weakness, foot drop and neck weakness are the main symptoms at onset, but ultimately the weakness usually involves the proximal compartment of both upper and lower limbs. Additional features include variable degrees of Achilles tendon contractures, spinal rigidity and muscle hypertrophy. Respiratory involvement often leads to requirement for non-invasive ventilation support.
Common junctional plaque protein. The membrane-associated plaques are architectural elements in an important strategic position to influence the arrangement and function of both the cytoskeleton and the cells within the tissue. The presence of plakoglobin in both the desmosomes and in the intermediate junctions suggests that it plays a central role in the structure and function of submembranous plaques. Acts as a substrate for VE-PTP and is required by it to stimulate VE-cadherin function in end
Cell junction, adherens junctionCell junction, desmosomeCytoplasm, cytoskeletonCell membraneCytoplasmCell junctionNucleusCell projection, axon
Naxos disease
An autosomal recessive disorder characterized by the association of diffuse non-epidermolytic palmoplantar keratoderma with woolly hair and cardiac abnormalities such as dilated cardiomyopathy and arrhythmogenic right ventricular dysplasia.
A component of desmosome cell-cell junctions which are required for positive regulation of cellular adhesion (PubMed:25208567). Regulates focal adhesion turnover resulting in changes in focal adhesion size, cell adhesion and cell spreading, potentially via transcriptional modulation of beta-integrins (PubMed:23884246). Required to maintain gingival epithelial barrier function (PubMed:34368962). Important component of the desmosome that is also required for localization of desmosome component pro
NucleusCell junction, desmosomeCell junctionCytoplasm
Arrhythmogenic right ventricular dysplasia, familial, 9
A congenital heart disease characterized by infiltration of adipose and fibrous tissue into the right ventricle and loss of myocardial cells, resulting in ventricular and supraventricular arrhythmias.
Variantes genéticas (ClinVar)
1.765 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
44 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Cardiomiopatia arritmogênica isolada hereditária
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Clinical Spectrum of Arrhythmogenic Entities in Spanish Children Carrying Deleterious SCN5A Variants.
Deleterious variants in SCN5A lead to a wide clinical spectrum that includes pathologies characterized by life-threatening cardiac events (CEs). In the pediatric population, early identification, management, and risk stratification of these pathologies are the main current challenges. This study analyzed a Spanish pediatric cohort (≤18 years) carrying rare SCN5A variants to explore genotype-phenotype correlations. A retrospective descriptive cohort study, including clinical, demographic, and genetic data of probands and their relatives, was conducted. Out of 100 children studied, 69 had definitively deleterious SCN5A variants (26 females, 38%; median age: 3 years, IQR 1-12). The main diagnoses were isolated Brugada syndrome (BrS) (31; 45%); isolated long QT syndrome type 3 (LQT3) (5; 7%); isolated progressive cardiac conduction disease (PCCD) (1; 2%); isolated familial atrial fibrillation (1; 2%); overlapping phenotypes (7; 10%) including: BrS-PCCD (2; 2.8%); BrS-LQT3 (1; 1.4%); premature ventricular contraction-dilated cardiomyopathy (1; 1.4%); BrS-LQT3-PCCD (1; 1.4%); BrS-PCCD-sick sinus syndrome (SSS) (1; 1.4%) and BrS-PCCD-SSS-familial atrial fibrillation (1; 1.4%). Of them, 13 (19%) patients presented with CEs (cardiogenic syncope, ventricular tachycardia/fibrillation, sudden cardiac arrest/death, and appropriate implantable cardio defibrillator shock). These findings underscore the utility of genetic testing for early diagnosis, risk stratification, and personalized management, enhancing preventive strategies for CE prevention in pediatrics.
Chemotherapy-Induced Arrhythmogenic Right Ventricular Cardiomyopathy.
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited disorder characterized by fibrofatty replacement of the right ventricular myocardium. Anthracycline-induced cardiotoxicity typically manifests as left ventricular dysfunction; however, we present a unique case of an acquired ARVC phenotype secondary to anthracycline chemotherapy exposure. A 74-year-old man developed recurrent exertional syncope 8 months after completing rituximab, cyclophosphamide, hydroxydaunorubicin (an anthracycline that is also known as doxorubicin), oncovin, and prednisone chemotherapy. He met 3 major 2010 Revised Task Force Criteria for definite ARVC, despite negative genetic testing and absence of family history. He underwent dual-chamber implantable cardioverter-defibrillator implantation with complete resolution of syncopal episodes. Recognition of acquired ARVC after anthracycline exposure broadens the spectrum of chemotherapy-related cardiomyopathies and calls for vigilance in patients with unexplained right ventricular dysfunction or arrhythmias after chemotherapy. Anthracyclines may cause isolated right ventricular dysfunction mimicking ARVC. Clinicians should maintain vigilance for right ventricular dysfunction or arrhythmias in patients after anthracycline therapy.
Isolated Nonischemic Left Ventricular Scar in Asymptomatic Athletes: Clinical Characteristics, Genetic Background, and Follow-Up.
Isolated nonischemic left ventricular scar (NILVS), identified by cardiac magnetic resonance, is an increasing finding in athletes and may be the substrate for life-threatening arrhythmias. Clinical significance in asymptomatic athletes is yet to be investigated. This study sought to describe the clinico-genetic profile and follow-up of asymptomatic athletes diagnosed with NILVS through preparticipation screening. We evaluated 40 athletes (90% males, 44 [range:33-52] years) including 9 elites, with isolated NILVS involving at least 2 segments, no previous major arrhythmic events, with available genetic testing and >1 year of follow-up. Data regarding electrocardiography, echocardiography, 24-hour Holter, exercise testing, and genetic analysis were collected. Follow-up assessed therapy, sport participation, and outcome. Electrocardiogram abnormalities were present in 48%, and all showed premature ventricular beats at exercise testing, mostly with right bundle branch/superior axis morphology. Left ventricular ejection fraction was normal or mildly reduced. Genetic testing or family screening was positive in 9 (23%). Athletes without familial/genetic background were older and declared higher cumulative years of sports activity. Over a median follow-up time of 23 months, 84% continued noncompetitive sport, mostly (73%) on beta-blocker therapy. Two major arrhythmic events occurred (resuscitated cardiac arrest and sustained ventricular tachycardia), both in athletes with a positive family history for NILVS, but negative genetic testing, and both during noncompetitive exercise. NILVS in asymptomatic athletes may carry arrhythmic risk even in the absence of previous symptoms or left ventricular dysfunction. Athletes with NILVS and no gene mutations/family history are older and with a higher past exercise volume.
Apremilast improves cardiomyocyte cohesion and arrhythmia in different models for arrhythmogenic cardiomyopathy.
Arrhythmogenic cardiomyopathy (ACM) is a genetically inherited desmosome heart disease leading to life-threatening arrhythmias and sudden cardiac death. Currently, ACM treatment paradigms are merely symptom targeting. Recently, apremilast was shown to stabilize keratinocyte adhesion in the desmosomal disease pemphigus vulgaris. Therefore, this study investigated whether apremilast can be a therapeutic option for ACM. Human induced pluripotent stem cells from a healthy control (hiPSC) and an ACM index patient (ACM-hiPSC) carrying a heterozygous desmoplakin (DSP) gene mutation (c.2854G > T, p.Glu952Ter), confirmed by whole exome sequencing (WES), were established. Cyclic-AMP ELISA, dissociation assay, immunostaining, and Western blotting analyses were performed in human iPSC-derived cardiomyocytes (hiPSC-CMs), murine HL-1 cardiomyocytes, and cardiac slices derived from wild-type (WT) mice, plakoglobin (PG, Jup) knockout (Jup-/-) (murine ACM model) or PG Serine 665 phosphodeficient (JUP-S665A) mice. Microelectrode array (MEA) analyses in ventricular cardiac slices and Langendorff heart perfusion were performed to analyze heart rate variability and arrhythmia. ACM-hiPSC derived cardiomyocytes (ACM-hiPSC-CMs) revealed a significant loss of cohesion, which was rescued by apremilast. Further, treatment with apremilast strengthened basal cardiomyocyte cohesion in HL-1 cells and WT murine cardiac slices, paralleled by phosphorylation of PG at Serine 665 in human and murine models. In HL-1 cells, apremilast in addition activated ERK1/2, inhibition of which abolished apremilast-enhanced cardiomyocyte cohesion. Further, dissociation assays in slice cultures from JUP-S665A and Jup-/- mice revealed that PG is crucial for apremilast-enhanced cardiomyocyte cohesion. In parallel to enhanced cell adhesion, MEA and Langendorff measurements from WT and Jup-/- mice demonstrated decreased heart rate variability and arrhythmia after apremilast treatment. Apremilast improves loss of cardiomyocyte cohesion, enhances localization of DSG2, and reduces arrhythmia in human and/or murine models of ACM ex vivo and in vitro, providing a novel treatment strategy for ACM by preserving desmosome function.
Hypercontractility and Oxidative Stress Drive Creatine Kinase Dysfunction in Hypertrophic Cardiomyopathy.
Hypertrophic cardiomyopathy (HCM) is a prevalent inherited cardiac disorder marked by left ventricular hypertrophy and hypercontractility. This excessive mechanical workload creates an energetic mismatch in which consumption exceeds production, leading to myocardial energy depletion. Although CK (creatine kinase) plays a key role in cardiac energy homeostasis, its involvement in HCM remains unclear. This study investigates how hypercontractility-driven mitochondrial stress and the resulting increase in mitochondrial H2O2 disrupt CK function in HCM. CK function was analyzed using myocardial left ventricular tissue from 92 patients with HCM (with and without pathogenic sarcomere variants) and 30 non-failing human controls. Myofilament and mitochondrial CK isoforms were measured using mRNA analysis, protein immunoblotting, enzyme activity assays, mass spectrometry, and redox-sensitive proteomics. To explore links between hypercontractility, mitochondrial reactive oxygen species, and CK dysfunction, we used isolated cardiomyocytes from wild-type, mitochondrial-targeted catalase-overexpressing, CK knockout (myofilament and mitochondrial CK deletion), HCM-associated Mybpc3 knock-in, and mito-roGFP2-Orp1 mouse models. We also tested the effects of the Ca2+ sensitizer EMD-57033, the CK inhibitor 1-fluoro-2,4-dinitrobenzene (DNFB), and the myosin inhibitor MYK-581, a mavacamten derivative. Our analysis revealed significant reductions in myofilament and mitochondrial CK protein levels, as well as CK activity, in myocardium of patients with HCM, primarily because of oxidative modifications of CK. In isolated mouse cardiomyocytes from wild-type and CK knockouts, hypercontractility induced by EMD-57033 elevated mitochondrial H2O2, causing cellular arrhythmias and CK inactivation. Hypercontractility-induced oxidative stress, arrhythmias, and CK dysfunction were also observed in Mybpc3 knock-in cardiomyocytes. Mitochondrial-targeted catalase-overexpressing mice with enhanced H2O2 scavenging were protected against H2O2-induced (EMD-57033-mediated) arrhythmias and CK dysfunction. MYK-581 treatment in Mybpc3 knock-in cardiomyocytes reduced hypercontractility, lowered H2O2 production and arrhythmias, and preserved CK function. CK inhibition using DNFB in wild-type cardiomyocytes elevated mitochondrial H2O2 levels and triggered cellular arrhythmias. This mitochondrial oxidation was independently confirmed in mito-roGFP2-Orp1 cardiomyocytes exposed to DNFB. Mitochondrial-targeted catalase-overexpressing mice were protected from DNFB-induced oxidative stress and arrhythmogenic events. This study reveals a mechanistic link between hypercontractility, mitochondrial reactive oxygen species, and CK dysfunction in HCM, perpetuating a cycle of energetic dysfunction. Targeting hypercontractility and oxidative stress through myosin inhibition offers a strategy to restore energy balance and reduce arrhythmic risk in HCM.
Publicações recentes
Peritoneal dialysis in congestive heart failure: reflections from a patient with arrhythmogenic right ventricular cardiomyopathy.
Arrhythmogenic Cardiomyopathy PKP2-Related: Clinical and Functional Characterization of a Pathogenic Variant Detected in Two Italian Families.
Sudden cardiac death, arrhythmogenic cardiomyopathy and intercalated disc pathology due to reduced filamin C protein levels: a matter of life and death.
Hyperactivation of ATF4/TGF-β1 signaling contributes to the progressive cardiac fibrosis in Arrhythmogenic cardiomyopathy caused by DSG2 Variant.
Cardiac sympathetic neurons are additional cells affected in genetically determined arrhythmogenic cardiomyopathy.
📚 EuropePMCmostrando 40
Chemotherapy-Induced Arrhythmogenic Right Ventricular Cardiomyopathy.
JACC. Case reportsClinical Spectrum of Arrhythmogenic Entities in Spanish Children Carrying Deleterious SCN5A Variants.
International journal of molecular sciencesArrhythmias across the tree of life: comparative insights for human electrophysiology.
Frontiers in cardiovascular medicineIsolated Nonischemic Left Ventricular Scar in Asymptomatic Athletes: Clinical Characteristics, Genetic Background, and Follow-Up.
JACC. Clinical electrophysiologyAtrial Dilated Cardiomyopathy: From Molecular Pathogenesis to Clinical Implications.
Journal of clinical medicineGenetic and Clinical Characterization of FLNC Variants in Chinese Patients with Cardiomyopathy.
Journal of cardiovascular development and diseaseRight Ventricular Outflow Tract Diameter for Event Prediction in Arrhythmogenic Right Ventricular Cardiomyopathy: Incremental Value Over Echocardiographic Free-Wall Strain.
JACC. Cardiovascular imagingEpicardial contributions to fibro-inflammatory signaling in a Pkp2-deficient arrhythmogenic cardiomyopathy model.
bioRxiv : the preprint server for biologyApremilast improves cardiomyocyte cohesion and arrhythmia in different models for arrhythmogenic cardiomyopathy.
Stem cell research & therapyHypercontractility and Oxidative Stress Drive Creatine Kinase Dysfunction in Hypertrophic Cardiomyopathy.
CirculationArrhythmogenic Cardiomyopathy with Biventricular Involvement and Right Ventricular Thrombosis: A Multi-modality Imaging Approach.
Heart views : the official journal of the Gulf Heart AssociationNPPA-Associated Atrial Dilated Cardiomyopathy: Genotypic and Phenotypic Insights From an Ultrarare Inherited Disorder.
JACC. Case reportsDiagnostic Criteria and Disease Staging for Desmoplakin Cardiomyopathy.
medRxiv : the preprint server for health sciencesArrhythmogenic Cardiomyopathy PKP2-Related: Clinical and Functional Characterization of a Pathogenic Variant Detected in Two Italian Families.
GenesAnalysis of effector/memory regulatory T cells from arrhythmogenic cardiomyopathy patients identified IL-32 as a novel player in ACM pathogenesis.
Cell death & diseaseHyperactivation of ATF4/TGF-β1 signaling contributes to the progressive cardiac fibrosis in Arrhythmogenic cardiomyopathy caused by DSG2 Variant.
BMC medicineExtracellular Kir2.1C122Y Mutant Upsets Kir2.1-PIP2 Bonds and Is Arrhythmogenic in Andersen-Tawil Syndrome.
Circulation researchArrhythmogenic Right Ventricular Cardiomyopathy Post-Mortem Assessment: A Systematic Review.
International journal of molecular sciencesIsolated JUP plakoglobin gene mutation with left ventricular fibrosis in familial arrhythmogenic right ventricular cardiomyopathy.
Journal of cardiovascular electrophysiologyDesmosomal protein degradation as an underlying cause of arrhythmogenic cardiomyopathy.
Science translational medicineConcealed Substrates in Brugada Syndrome: Isolated Channelopathy or Associated Cardiomyopathy?
GenesContinuous-flow left ventricular assist device treatment for arrhythmogenic right ventricular cardiomyopathy complicated by advanced biventricular failure - University of Tokyo experiences.
Frontiers in cardiovascular medicineIdentification of a novel variant in N-cadherin associated with dilated cardiomyopathy.
Frontiers in medicineProgressive Reduction in Right Ventricular Contractile Function Attributable to Altered Actin Expression in an Aging Mouse Model of Arrhythmogenic Cardiomyopathy.
CirculationNovel plasma biomarkers predicting biventricular involvement in arrhythmogenic right ventricular cardiomyopathy.
American heart journalA Case of Peripartum Ventricular Tachycardia due to Arrhythmogenic Right Ventricular Dysplasia.
South Dakota medicine : the journal of the South Dakota State Medical AssociationCardiac magnetic resonance in patients with ARVC and family members: the potential role of native T1 mapping.
The international journal of cardiovascular imagingCardiovascular Magnetic Resonance and Sport Cardiology: a Growing Role in Clinical Dilemmas.
Journal of cardiovascular translational researchOvercoming challenges in the management of arrhythmogenic right ventricular cardiomyopathy.
Kardiologia polskaFamilial screening in case of acute myocarditis reveals inherited arrhythmogenic left ventricular cardiomyopathies.
ESC heart failureThe evolution of gene-guided management of inherited arrhythmia syndromes: Peering beyond monogenic paradigms towards comprehensive genomic risk scores.
Journal of cardiovascular electrophysiologyG790del mutation in DSC2 alone is insufficient to develop the pathogenesis of ARVC in a mouse model.
Biochemistry and biophysics reportsSudden Death and Left Ventricular Involvement in Arrhythmogenic Cardiomyopathy.
CirculationAtrial fibrillation in patients with inherited cardiomyopathies.
Europace : European pacing, arrhythmias, and cardiac electrophysiology : journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of CardiologyNovel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia.
CirculationArrhythmogenic Right Ventricular Dysplasia: An Under-recognized Form of Inherited Cardiomyopathy.
Reviews in cardiovascular medicineThe Role of Genetic Testing in the Identification of Young Athletes with Inherited Primitive Cardiac Disorders at Risk of Exercise Sudden Death.
Frontiers in cardiovascular medicineCombination of palmoplantar keratoderma and hair shaft anomalies, the warning signal of severe arrhythmogenic cardiomyopathy: a systematic review on genetic desmosomal diseases.
Journal of medical geneticsRisk Stratification in Hypertrophic Cardiomyopathy.
European cardiologyArrhythmogenic right ventricular cardiomyopathy. Contribution of cardiac magnetic resonance imaging to the diagnosis.
HerzAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Cardiomiopatia arritmogênica isolada hereditária.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Cardiomiopatia arritmogênica isolada hereditária
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Clinical Spectrum of Arrhythmogenic Entities in Spanish Children Carrying Deleterious SCN5A Variants.
- Chemotherapy-Induced Arrhythmogenic Right Ventricular Cardiomyopathy.
- Isolated Nonischemic Left Ventricular Scar in Asymptomatic Athletes: Clinical Characteristics, Genetic Background, and Follow-Up.
- Apremilast improves cardiomyocyte cohesion and arrhythmia in different models for arrhythmogenic cardiomyopathy.
- Hypercontractility and Oxidative Stress Drive Creatine Kinase Dysfunction in Hypertrophic Cardiomyopathy.
- Peritoneal dialysis in congestive heart failure: reflections from a patient with arrhythmogenic right ventricular cardiomyopathy.
- Arrhythmogenic Cardiomyopathy PKP2-Related: Clinical and Functional Characterization of a Pathogenic Variant Detected in Two Italian Families.
- Sudden cardiac death, arrhythmogenic cardiomyopathy and intercalated disc pathology due to reduced filamin C protein levels: a matter of life and death.
- Hyperactivation of ATF4/TGF-β1 signaling contributes to the progressive cardiac fibrosis in Arrhythmogenic cardiomyopathy caused by DSG2 Variant.
- Cardiac sympathetic neurons are additional cells affected in genetically determined arrhythmogenic cardiomyopathy.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:217656(Orphanet)
- MONDO:0016342(MONDO)
- GARD:17129(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q56013813(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
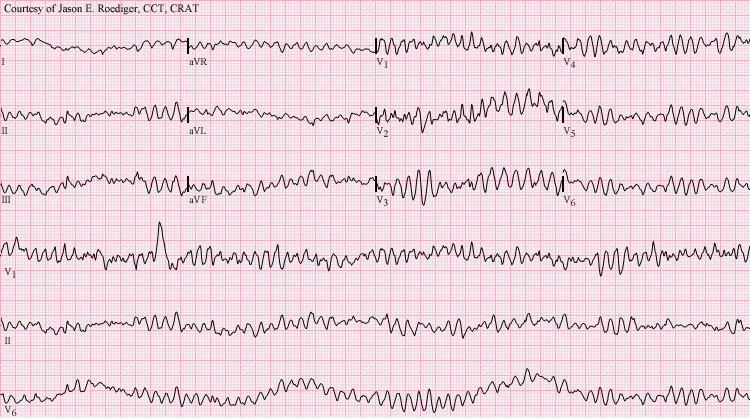
Cardiomiopatia arritmogênica isolada hereditária
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub