Doença neurológica que afeta as células nervosas responsáveis pelos movimentos.
Introdução
O que você precisa saber de cara
Doença neurológica que afeta as células nervosas responsáveis pelos movimentos.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 290 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 666 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
69 genes identificados com associação a esta condição.
Catalyzes the ATP-dependent ligation of glycine to the 3'-end of its cognate tRNA, via the formation of an aminoacyl-adenylate intermediate (Gly-AMP) (PubMed:17544401, PubMed:24898252, PubMed:28675565). Also produces diadenosine tetraphosphate (Ap4A), a universal pleiotropic signaling molecule needed for cell regulation pathways, by direct condensation of 2 ATPs. Thereby, may play a special role in Ap4A homeostasis (PubMed:19710017)
CytoplasmCell projection, axonSecretedSecreted, extracellular exosomeMitochondrion
Charcot-Marie-Tooth disease, axonal, type 2D
A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy.
Has low activity towards the organophosphate paraxon and aromatic carboxylic acid esters. Rapidly hydrolyzes lactones such as statin prodrugs (e.g. lovastatin). Hydrolyzes aromatic lactones and 5- or 6-member ring lactones with aliphatic substituents but not simple lactones or those with polar substituents
Secreted, extracellular space
Plays an important role in the maintenance of the Golgi complex, in membrane trafficking, in exocytosis, through its interaction with myosin VI and Rab8 (PubMed:27534431). Links myosin VI to the Golgi complex and plays an important role in Golgi ribbon formation (PubMed:27534431). Plays a role in the activation of innate immune response during viral infection. Mechanistically, recruits TBK1 at the Golgi apparatus, promoting its trans-phosphorylation after RLR or TLR3 stimulation (PubMed:27538435
Cytoplasm, perinuclear regionGolgi apparatusGolgi apparatus, trans-Golgi networkCytoplasmic vesicle, autophagosomeCytoplasmic vesicleRecycling endosome
Glaucoma 1, open angle, E
A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place.
Transcriptional coactivator for steroid receptors and nuclear receptors (PubMed:10713165, PubMed:20005308, PubMed:21376232, PubMed:28363985, PubMed:32433991). Greatly increases the transcriptional activity of PPARG and thyroid hormone receptor on the uncoupling protein promoter (PubMed:10713165, PubMed:20005308, PubMed:21376232). Can regulate key mitochondrial genes that contribute to the program of adaptive thermogenesis (PubMed:10713165, PubMed:20005308, PubMed:21376232). Plays an essential ro
NucleusNucleus, PML bodyCytoplasm
Functions as a guanine exchange factor (GEF) for RAB26 and thus regulates autophagy of synaptic vesicles in axon terminal of motoneurons (By similarity). Involved in the control of neuronal cell differentiation (PubMed:11704860). Plays a role in angiogenesis through regulation of endothelial cells chemotaxis. Also affects the migration, adhesion, and matrix/bone degradation in macrophages and osteoclasts (PubMed:23777631)
CytoplasmCytoplasm, perinuclear regionCell membraneCell junctionCell projection, lamellipodium
Neuronopathy, distal hereditary motor, autosomal recessive 4
A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR4 is characterized by childhood onset, generalized muscle weakness and atrophy with denervation and normal sensation. Bulbar symptoms and pyramidal signs are absent.
Plasma membrane transporter mediating the uptake by cells of the water soluble vitamin B2/riboflavin that plays a key role in biochemical oxidation-reduction reactions of the carbohydrate, lipid, and amino acid metabolism (PubMed:20463145, PubMed:22273710, PubMed:24264046, PubMed:27702554). Humans are unable to synthesize vitamin B2/riboflavin and must obtain it via intestinal absorption (PubMed:20463145)
Apical cell membraneCell membraneNucleus membraneCytoplasm
Brown-Vialetto-Van Laere syndrome 1
A rare neurologic disorder characterized by sensorineural hearing loss and a variety of cranial nerve palsies, which develop over a relatively short period of time in a previously healthy individual. Sensorineural hearing loss may precede the neurological signs. The course is invariably progressive, but the rate of decline is variable within and between families. With disease evolution, long tract signs, lower motor neuron signs, cerebellar ataxia and lower cranial nerve (III-VI) palsies develop, giving rise to a complex picture resembling amyotrophic lateral sclerosis. Diaphragmatic weakness and respiratory compromise are some of the most distressing features, leading to recurrent chest infections and respiratory failure, which are often the cause of patients' demise.
Catalytic component of the m-AAA protease, a protease that plays a key role in proteostasis of inner mitochondrial membrane proteins, and which is essential for axonal and neuron development (PubMed:11549317, PubMed:28396416, PubMed:31097542, PubMed:9635427). SPG7 possesses both ATPase and protease activities: the ATPase activity is required to unfold substrates, threading them into the internal proteolytic cavity for hydrolysis into small peptide fragments (By similarity). The m-AAA protease ex
Mitochondrion inner membrane
Spastic paraplegia 7, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG7 is a complex form. Additional clinical features are cerebellar syndrome, supranuclear palsy, and cognitive impairment, particularly disturbance of attention and executive functions.
Metallocarboxypeptidase that mediates protein deglutamylation of tubulin and non-tubulin target proteins (PubMed:22170066, PubMed:24022482, PubMed:30420557). Catalyzes the removal of polyglutamate side chains present on the gamma-carboxyl group of glutamate residues within the C-terminal tail of alpha- and beta-tubulin (PubMed:22170066, PubMed:24022482, PubMed:30420557). Specifically cleaves tubulin long-side-chains, while it is not able to remove the branching point glutamate (PubMed:24022482).
CytoplasmCytoplasm, cytosolNucleusMitochondrion
Neurodegeneration, childhood-onset, with cerebellar atrophy
An autosomal recessive disorder characterized by early onset of progressive neurodegeneration affecting the central and peripheral nervous systems. Clinical features include global developmental delay, impaired intellectual development, poor or absent speech, and motor abnormalities. Brain imaging shows cerebellar atrophy. Death in childhood may occur.
Tubulin-folding protein; involved in the second step of the tubulin folding pathway and in the regulation of tubulin heterodimer dissociation. Required for correct organization of microtubule cytoskeleton and mitotic splindle, and maintenance of the neuronal microtubule network
CytoplasmCytoplasm, cytoskeleton
Hypoparathyroidism-retardation-dysmorphism syndrome
An autosomal recessive multisystem disorder characterized by hypoparathyroidism, intrauterine and postnatal growth retardation, psychomotor retardation, epilepsy, microcephaly, and facial dysmorphism.
Destroys radicals which are normally produced within the cells and which are toxic to biological systems (PubMed:24140062). Catalyzes the oxidation of hydrogen sulfide (H2S) to sulfate, playing an important role in detoxifying H2S and limiting the accumulation of reactive sulfur species (RSS) such as persulfides and polysulfides (PubMed:36630448)
CytoplasmNucleus
Amyotrophic lateral sclerosis 1
A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases.
Plays a role in cilia formation and/or maintenance (By similarity). Plays a role in the regulation of cell morphology and cytoskeletal organization (PubMed:21834987). Involved in DNA damage repair (PubMed:26290490)
MitochondrionCytoplasm, cytoskeleton, cilium basal bodyCell projection, cilium, photoreceptor outer segmentCytoplasm
Retinal dystrophy with or without macular staphyloma
An ocular disorder characterized by decreased vision which worsen over time, and dystrophic changes in the retina, such as retinal pigment epithelium mottling and vessel narrowing. Macular staphyloma, without high myopia, is present in some patients.
In vitro, catalyzes the transfer of a galactose residue from UDP-galactose onto GalNAc and GlcNAc structures
Golgi apparatus membrane
Involved in the packaging of pre-mRNA into hnRNP particles, transport of poly(A) mRNA from the nucleus to the cytoplasm and modulation of splice site selection (PubMed:17371836). Plays a role in the splicing of pyruvate kinase PKM by binding repressively to sequences flanking PKM exon 9, inhibiting exon 9 inclusion and resulting in exon 10 inclusion and production of the PKM M2 isoform (PubMed:20010808). Binds to the IRES and thereby inhibits the translation of the apoptosis protease activating
NucleusCytoplasm
Inclusion body myopathy with early-onset Paget disease with or without frontotemporal dementia 3
An autosomal dominant disease characterized by disabling muscle weakness clinically resembling to limb girdle muscular dystrophy, osteolytic bone lesions consistent with Paget disease, and premature frontotemporal dementia. Clinical features show incomplete penetrance.
Catalyzes the oxidative deamination of primary amines to the corresponding aldehydes with the concomitant production of hydrogen peroxide and ammonia (PubMed:12072962, PubMed:19764817, PubMed:239684, PubMed:8144586). Its preferred substrates are the diamines histamine and 1-methylhistamine and it could therefore play a role in allergic and immune responses (PubMed:12072962). Has a broad specificity for diamines and can also act on cadaverine and putrescine, two products of amino acid catabolism
Secreted, extracellular spaceCell membrane
Phosphorylates serines and threonines, but also appears to possess tyrosine kinase activity (PubMed:20230784). Involved in DNA damage checkpoint control and for proper DNA damage repair (PubMed:20230784). In response to injury that includes DNA damage, NEK1 phosphorylates VDAC1 to limit mitochondrial cell death (PubMed:20230784). May be implicated in the control of meiosis (By similarity). Involved in cilium assembly (PubMed:21211617)
NucleusCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm
Short-rib thoracic dysplasia 6 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Required for the export of mRNAs containing poly(A) tails from the nucleus into the cytoplasm. May be involved in the terminal step of the mRNA transport through the nuclear pore complex (NPC)
NucleusCytoplasmNucleus, nuclear pore complex
Lethal congenital contracture syndrome 1
A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS1 patients manifest early fetal hydrops and akinesia, micrognathia, pulmonary hypoplasia, pterygia, and multiple joint contractures. It leads to prenatal death.
Capable of hydrolyzing lactones and a number of aromatic carboxylic acid esters. Has antioxidant activity. Is not associated with high density lipoprotein. Prevents LDL lipid peroxidation, reverses the oxidation of mildly oxidized LDL, and inhibits the ability of MM-LDL to induce monocyte chemotaxis
Membrane
The TFIID basal transcription factor complex plays a major role in the initiation of RNA polymerase II (Pol II)-dependent transcription (PubMed:33795473). TFIID recognizes and binds promoters with or without a TATA box via its subunit TBP, a TATA-box-binding protein, and promotes assembly of the pre-initiation complex (PIC) (PubMed:33795473). The TFIID complex consists of TBP and TBP-associated factors (TAFs), including TAF1, TAF2, TAF3, TAF4, TAF5, TAF6, TAF7, TAF8, TAF9, TAF10, TAF11, TAF12 an
Nucleus
Transmembrane protein of the mitochondrial outer membrane that controls mitochondrial organization (PubMed:26168012, PubMed:27390132, PubMed:27543974). May regulate the assembly of the MICOS (mitochondrial contact site and cristae organizing system) complex which is essential to the biogenesis and dynamics of mitochondrial cristae, the inwards folds of the inner mitochondrial membrane (PubMed:27390132). Through its interaction with the EMC (endoplasmic reticulum membrane protein complex), could
Mitochondrion outer membrane
Neuropathy, hereditary motor and sensory, 6B, with optic atrophy
An autosomal recessive neurologic disorder characterized by early-onset optic atrophy, progressive visual loss, and peripheral sensorimotor neuropathy manifesting as axonal Charcot-Marie-Tooth disease, with variable age at onset and severity. Charcot-Marie-Tooth disease is a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. It is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies and primary peripheral axonal neuropathies. Peripheral axonal neuropathies are characterized by signs of axonal regeneration in the absence of obvious myelin alterations, and normal or slightly reduced nerve conduction velocities.
RNA-binding protein that is involved in various steps of RNA biogenesis and processing (PubMed:23519609). Preferentially binds, via its two RNA recognition motifs RRM1 and RRM2, to GU-repeats on RNA molecules predominantly localized within long introns and in the 3'UTR of mRNAs (PubMed:23519609, PubMed:24240615, PubMed:24464995). In turn, regulates the splicing of many non-coding and protein-coding RNAs including proteins involved in neuronal survival, as well as mRNAs that encode proteins relev
NucleusCytoplasmCytoplasm, Stress granuleMitochondrion
Amyotrophic lateral sclerosis 10
A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases.
Serine/threonine kinase that plays an essential role in regulating inflammatory responses to foreign agents (PubMed:10581243, PubMed:11839743, PubMed:12692549, PubMed:12702806, PubMed:14703513, PubMed:15367631, PubMed:15485837, PubMed:18583960, PubMed:21138416, PubMed:23453971, PubMed:23453972, PubMed:23746807, PubMed:25636800, PubMed:26611359, PubMed:32404352, PubMed:34363755, PubMed:32298923). Following activation of toll-like receptors by viral or bacterial components, associates with TRAF3 a
Cytoplasm
Glaucoma 1, open angle, P
A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place. GLC1P is characterized by early onset, thin central corneas and low intraocular pressure.
Essential for retina photoreceptor outer segment disk morphogenesis, may also play a role with ROM1 in the maintenance of outer segment disk structure (By similarity). Required for the maintenance of retinal outer nuclear layer thickness (By similarity). Required for the correct development and organization of the photoreceptor inner segment (By similarity)
MembraneCell projection, cilium, photoreceptor outer segmentPhotoreceptor inner segment
Retinitis pigmentosa 7
A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well.
Component of the ERLIN1/ERLIN2 complex which mediates the endoplasmic reticulum-associated degradation (ERAD) of inositol 1,4,5-trisphosphate receptors (IP3Rs) such as ITPR1 (PubMed:17502376, PubMed:19240031). Promotes sterol-accelerated ERAD of HMGCR probably implicating an AMFR/gp78-containing ubiquitin ligase complex (PubMed:21343306). Involved in regulation of cellular cholesterol homeostasis by regulation the SREBP signaling pathway. May promote ER retention of the SCAP-SREBF complex (PubMe
Endoplasmic reticulum membrane
Spastic paraplegia 18B, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG18B is a severe form with onset in early childhood. Most affected individuals have severe psychomotor retardation. Some may develop significant joint contractures.
DNA/RNA-binding protein that plays a role in various cellular processes such as transcription regulation, RNA splicing, RNA transport, DNA repair and damage response (PubMed:27731383). Binds to ssRNA containing the consensus sequence 5'-AGGUAA-3' (PubMed:21256132). Binds to nascent pre-mRNAs and acts as a molecular mediator between RNA polymerase II and U1 small nuclear ribonucleoprotein thereby coupling transcription and splicing (PubMed:26124092). Also binds its own pre-mRNA and autoregulates
Nucleus
The SMN complex catalyzes the assembly of small nuclear ribonucleoproteins (snRNPs), the building blocks of the spliceosome, and thereby plays an important role in the splicing of cellular pre-mRNAs (PubMed:18984161, PubMed:9845364). Most spliceosomal snRNPs contain a common set of Sm proteins SNRPB, SNRPD1, SNRPD2, SNRPD3, SNRPE, SNRPF and SNRPG that assemble in a heptameric protein ring on the Sm site of the small nuclear RNA to form the core snRNP (Sm core) (PubMed:18984161). In the cytosol,
Nucleus, gemNucleus, Cajal bodyCytoplasmCytoplasmic granulePerikaryonCell projection, neuron projectionCell projection, axonCytoplasm, myofibril, sarcomere, Z line
Spinal muscular atrophy 1
A form of spinal muscular atrophy, a group of neuromuscular disorder characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. Autosomal recessive forms are classified according to the age of onset, the maximum muscular activity achieved, and survivorship. The severity of the disease is mainly determined by the copy number of SMN2, a copy gene which predominantly produces exon 7-skipped transcripts and only low amount of full-length transcripts that encode for a protein identical to SMN1. Only about 4% of SMA patients bear one SMN1 copy with an intragenic mutation. SMA1 is a severe form, with onset before 6 months of age. SMA1 patients never achieve the ability to sit.
Non-catalytic component of the RNA exosome complex which has 3'->5' exoribonuclease activity and participates in a multitude of cellular RNA processing and degradation events. In the nucleus, the RNA exosome complex is involved in proper maturation of stable RNA species such as rRNA, snRNA and snoRNA, in the elimination of RNA processing by-products and non-coding 'pervasive' transcripts, such as antisense RNA species and promoter-upstream transcripts (PROMPTs), and of mRNAs with processing defe
CytoplasmNucleusNucleus, nucleolusNucleus, nucleoplasm
Pontocerebellar hypoplasia 1D
An autosomal recessive neurologic disorder with onset at birth or in infancy, and characterized by progressive axonal motor neuronopathy, severe generalized hypotonia, respiratory insufficiency, and cerebellar atrophy. Death in childhood may occur.
Anti-apoptotic protein which acts by inhibiting the activities of CASP3, CASP7 and CASP9. Can inhibit the autocleavage of pro-CASP9 and cleavage of pro-CASP3 by CASP9. Capable of inhibiting CASP9 autoproteolysis at 'Asp-315' and decreasing the rate of auto proteolysis at 'Asp-330'. Acts as a mediator of neuronal survival in pathological conditions. Prevents motor-neuron apoptosis induced by a variety of signals. Possible role in the prevention of spinal muscular atrophy that seems to be caused b
ATP-dependent 5'->3' DNA/RNA helicase that preferentially unwinds RNA substrates over DNA, playing a crucial role in resolving R-loops and promoting transcription termination (PubMed:36864660). Plays a role in transcription regulation by its ability to modulate RNA Polymerase II (Pol II) binding to chromatin and through its interaction with proteins involved in transcription (PubMed:19515850, PubMed:21700224). Contributes to the mRNA splicing efficiency and splice site selection (PubMed:19515850
NucleusNucleus, nucleoplasmNucleus, nucleolusCytoplasmChromosomeChromosome, telomereCell projection, axonCell projection, growth cone
Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 2
A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAN2 is an autosomal recessive form associated with peripheral neuropathy and elevated serum alpha-fetoprotein, immunoglobulins and, less commonly, creatine kinase levels. Some SCAN2 patients manifest oculomotor apraxia.
Deubiquitinase that specifically cleaves 'Lys-63'- and linear 'Met-1'-linked polyubiquitin chains and is involved in NF-kappa-B activation and TNF-induced necroptosis (PubMed:18313383, PubMed:18636086, PubMed:26670046, PubMed:26997266, PubMed:27458237, PubMed:27591049, PubMed:27746020, PubMed:29291351, PubMed:32185393). Negatively regulates NF-kappa-B activation by deubiquitinating upstream signaling factors (PubMed:12917689, PubMed:12917691, PubMed:32185393). Contributes to the regulation of ce
CytoplasmCytoplasm, perinuclear regionCytoplasm, cytoskeletonCell membraneCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, spindleCytoplasm, cytoskeleton, cilium basal body
Cylindromatosis, familial
A disorder characterized by multiple skin tumors that develop from skin appendages, such as hair follicles and sweat glands. Affected individuals typically develop large numbers of tumors called cylindromas that arise predominantly in hairy parts of the body with approximately 90% on the head and neck. In severely affected individuals, cylindromas may combine into a confluent mass which may ulcerate or become infected (turban tumor syndrome). Individuals with familial cylindromatosis occasionally develop other types of tumors including spiradenomas that begin in sweat glands, and trichoepitheliomas arising from hair follicles.
Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-depend
Nucleus inner membraneNucleus outer membraneNucleus envelopeCytoplasmic vesicleEndoplasmic reticulum membraneMembraneLipid dropletCell junctionCell membraneCell projection, growth conePostsynaptic density membrane
Amyotrophic lateral sclerosis 16, juvenile
A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases.
Molecular adapter required for selective macroautophagy (aggrephagy) by acting as a bridge between polyubiquitinated proteins and autophagosomes (PubMed:15340068, PubMed:15953362, PubMed:16286508, PubMed:17580304, PubMed:20168092, PubMed:22017874, PubMed:22622177, PubMed:24128730, PubMed:28404643, PubMed:29343546, PubMed:29507397, PubMed:31857589, PubMed:33509017, PubMed:34471133, PubMed:34893540, PubMed:35831301, PubMed:37306101, PubMed:37802024). Promotes the recruitment of ubiquitinated cargo
Cytoplasmic vesicle, autophagosomePreautophagosomal structureCytoplasm, cytosolNucleus, PML bodyLate endosomeLysosomeNucleusEndoplasmic reticulumCytoplasm, myofibril, sarcomere
Paget disease of bone 3
A disorder of bone remodeling characterized by increased bone turnover affecting one or more sites throughout the skeleton, primarily the axial skeleton. Osteoclastic overactivity followed by compensatory osteoblastic activity leads to a structurally disorganized mosaic of bone (woven bone), which is mechanically weaker, larger, less compact, more vascular, and more susceptible to fracture than normal adult lamellar bone.
Plays a role in DNA damage repair as component of the ASCC complex (PubMed:29997253). Part of the ASC-1 complex that enhances NF-kappa-B, SRF and AP1 transactivation (PubMed:12077347). In cells responding to gastrin-activated paracrine signals, it is involved in the induction of SERPINB2 expression by gastrin. May also play a role in the development of neuromuscular junction
NucleusNucleus speckle
Barrett esophagus
A condition characterized by a metaplastic change in which normal esophageal squamous epithelium is replaced by a columnar and intestinal-type epithelium. Patients with Barrett esophagus have an increased risk of esophageal adenocarcinoma. The main cause of Barrett esophagus is gastroesophageal reflux. The retrograde movement of acid and bile salts from the stomach into the esophagus causes prolonged injury to the esophageal epithelium and induces chronic esophagitis, which in turn is believed to trigger the pathologic changes.
Non-catalytic subunit of the tRNA-splicing endonuclease complex, a complex responsible for identification and cleavage of the splice sites in pre-tRNA. It cleaves pre-tRNA at the 5' and 3' splice sites to release the intron. The products are an intron and two tRNA half-molecules bearing 2',3' cyclic phosphate and 5'-OH termini. There are no conserved sequences at the splice sites, but the intron is invariably located at the same site in the gene, placing the splice sites an invariant distance fr
NucleusNucleus, nucleolus
Pontocerebellar hypoplasia 4
A disorder characterized by an abnormally small cerebellum and brainstem, severe neonatal encephalopathy, microcephaly, myoclonus and muscular hypertonia. There is a severe inferior olivary and pontine neuronal loss and a diffuse white matter gliosis.
Involved in the chaperone-assisted selective autophagy (CASA), a crucial process for protein quality control, particularly in mechanical strained cells and tissues such as muscle. Displays temperature-dependent chaperone activity
CytoplasmNucleus
Neuronopathy, distal hereditary motor, autosomal dominant 2
A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs.
Endoplasmic reticulum (ER)-anchored protein that mediates the formation of contact sites between the ER and endosomes via interaction with FFAT motif-containing proteins such as STARD3 or WDR44 (PubMed:32344433, PubMed:33124732). Interacts with STARD3 in a FFAT motif phosphorylation dependent manner (PubMed:33124732). Via interaction with WDR44 participates in neosynthesized protein export (PubMed:32344433). Participates in the endoplasmic reticulum unfolded protein response (UPR) by inducing ER
Endoplasmic reticulum membrane
Amyotrophic lateral sclerosis 8
A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases.
Catalyzes the first step in ubiquitin conjugation to mark cellular proteins for degradation through the ubiquitin-proteasome system (PubMed:1447181, PubMed:1606621, PubMed:33108101). Activates ubiquitin by first adenylating its C-terminal glycine residue with ATP, and thereafter linking this residue to the side chain of a cysteine residue in E1, yielding a ubiquitin-E1 thioester and free AMP (PubMed:1447181). Essential for the formation of radiation-induced foci, timely DNA repair and for respon
CytoplasmMitochondrionNucleus
Spinal muscular atrophy X-linked 2
A lethal infantile form of spinal muscular atrophy, a neuromuscular disorder characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. Clinical features include hypotonia, areflexia, and multiple congenital contractures.
Muscle-specific type III intermediate filament essential for proper muscular structure and function. Plays a crucial role in maintaining the structure of sarcomeres, inter-connecting the Z-disks and forming the myofibrils, linking them not only to the sarcolemmal cytoskeleton, but also to the nucleus and mitochondria, thus providing strength for the muscle fiber during activity (PubMed:25358400). In adult striated muscle they form a fibrous network connecting myofibrils to each other and to the
Cytoplasm, myofibril, sarcomere, Z lineCytoplasmCell membrane, sarcolemmaNucleusCell tipNucleus envelope
Myopathy, myofibrillar, 1
A form of myofibrillar myopathy, a group of chronic neuromuscular disorders characterized at ultrastructural level by disintegration of the sarcomeric Z disk and myofibrils, and replacement of the normal myofibrillar markings by small dense granules, or larger hyaline masses, or amorphous material. MFM1 is characterized by skeletal muscle weakness associated with cardiac conduction blocks, arrhythmias, restrictive heart failure, and accumulation of desmin-reactive deposits in cardiac and skeletal muscle cells.
Cytoplasmic dynein 1 acts as a motor for the intracellular retrograde motility of vesicles and organelles along microtubules. Dynein has ATPase activity; the force-producing power stroke is thought to occur on release of ADP. Plays a role in mitotic spindle assembly and metaphase plate congression (PubMed:27462074)
Cytoplasm, cytoskeleton
Charcot-Marie-Tooth disease, axonal, type 2O
An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy.
Lysosomal ceramidase that hydrolyzes sphingolipid ceramides into sphingosine and free fatty acids at acidic pH (PubMed:10610716, PubMed:11451951, PubMed:15655246, PubMed:26898341, PubMed:36752535, PubMed:7744740, PubMed:7852294). Ceramides, sphingosine, and its phosphorylated form sphingosine-1-phosphate are bioactive lipids that mediate cellular signaling pathways regulating several biological processes including cell proliferation, apoptosis and differentiation (PubMed:10610716). Has a higher
LysosomeSecretedNucleusCytoplasm
Farber lipogranulomatosis
An autosomal recessive lysosomal storage disorder characterized by subcutaneous lipid-loaded nodules, excruciating pain in the joints and extremities, and marked accumulation of ceramide in lysosomes. Disease severity is variable. The most severe disease subtype is a rare neonatal form with death occurring before 1 year of age.
Serine/threonine kinase involved in the regulation of key cellular processes including the cell cycle, nuclear condensation, transcription regulation, and DNA damage response (PubMed:14645249, PubMed:18617507, PubMed:19103756, PubMed:33076429). Controls chromatin organization and remodeling by mediating phosphorylation of histone H3 on 'Thr-4' and histone H2AX (H2aXT4ph) (PubMed:31527692, PubMed:37179361). It also phosphorylates KAT5 in response to DNA damage, promoting KAT5 association with chr
NucleusCytoplasmNucleus, Cajal body
Pontocerebellar hypoplasia 1A
A form of pontocerebellar hypoplasia, a disorder characterized by structural defects of the pons and cerebellum, evident upon brain imaging. PCH1A is an autosomal recessive form characterized by an abnormally small cerebellum and brainstem, central and peripheral motor dysfunction from birth, gliosis and spinal cord anterior horn cells degeneration resembling infantile spinal muscular atrophy. Additional features include muscle hypotonia, congenital contractures and respiratory insufficiency that is evident at birth.
Substrate recognition component of a SCF (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complex which mediates the ubiquitination and subsequent proteasomal degradation of PDCD1/PD-1, thereby regulating T-cells-mediated immunity (PubMed:30487606). Required for anti-tumor activity of T-cells by promoting the degradation of PDCD1/PD-1; the PDCD1-mediated inhibitory pathway being exploited by tumors to attenuate anti-tumor immunity and facilitate tumor survival (PubMed:30487606). May indirec
Cytoplasm, cytosolNucleus
Neuronopathy, distal hereditary motor, autosomal dominant 6
A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs.
ATP-driven copper (Cu(+)) ion pump that plays an important role in intracellular copper ion homeostasis (PubMed:10419525, PubMed:11092760, PubMed:28389643). Within a catalytic cycle, acquires Cu(+) ion from donor protein on the cytoplasmic side of the membrane and delivers it to acceptor protein on the lumenal side. The transfer of Cu(+) ion across the membrane is coupled to ATP hydrolysis and is associated with a transient phosphorylation that shifts the pump conformation from inward-facing to
Golgi apparatus, trans-Golgi network membraneCell membraneMelanosome membraneEarly endosome membraneCell projection, axonCell projection, dendritePostsynaptic densityCytoplasm, cytosolEndoplasmic reticulum
Menkes disease
An X-linked recessive disorder of copper metabolism characterized by generalized copper deficiency. MNKD results in progressive neurodegeneration and connective-tissue disturbances: focal cerebral and cerebellar degeneration, early growth retardation, peculiar hair, hypopigmentation, cutis laxa, vascular complications and death in early childhood. The clinical features result from the dysfunction of several copper-dependent enzymes. A mild form of the disease has been described, in which cerebellar ataxia and moderate developmental delay predominate.
May act as a GTPase regulator. Controls survival and growth of spinal motoneurons (By similarity)
Amyotrophic lateral sclerosis 2
A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases.
Small heat shock protein which functions as a molecular chaperone probably maintaining denatured proteins in a folding-competent state (PubMed:10383393, PubMed:20178975). Plays a role in stress resistance and actin organization (PubMed:19166925). Through its molecular chaperone activity may regulate numerous biological processes including the phosphorylation and the axonal transport of neurofilament proteins (PubMed:23728742)
CytoplasmNucleusCytoplasm, cytoskeleton, spindle
Charcot-Marie-Tooth disease, axonal, type 2F
A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. Onset of Charcot-Marie-Tooth disease type 2F is between 15 and 25 years with muscle weakness and atrophy usually beginning in feet and legs (peroneal distribution). Upper limb involvement occurs later.
Non-catalytic subunit of the tRNA-splicing endonuclease complex, a complex responsible for identification and cleavage of the splice sites in pre-tRNA. It cleaves pre-tRNA at the 5' and 3' splice sites to release the intron. The products are an intron and two tRNA half-molecules bearing 2',3' cyclic phosphate and 5'-OH termini (PubMed:15109492, PubMed:27392077). There are no conserved sequences at the splice sites, but the intron is invariably located at the same site in the gene, placing the sp
NucleusNucleus, nucleolus
Pontocerebellar hypoplasia 2F
A neurodevelopmental disorder characterized by progressive microcephaly, cognitive and motor delay, poor or absent speech, seizures, and spasticity. PCH2F inheritance is autosomal recessive.
Non-catalytic component of the RNA exosome complex which has 3'->5' exoribonuclease activity and participates in a multitude of cellular RNA processing and degradation events. In the nucleus, the RNA exosome complex is involved in proper maturation of stable RNA species such as rRNA, snRNA and snoRNA, in the elimination of RNA processing by-products and non-coding 'pervasive' transcripts, such as antisense RNA species and promoter-upstream transcripts (PROMPTs), and of mRNAs with processing defe
CytoplasmNucleusNucleus, nucleolus
Pontocerebellar hypoplasia 1C
A severe autosomal recessive neurodegenerative disease characterized by cerebellar and corpus callosum hypoplasia, abnormal myelination of the central nervous system, and spinal motor neuron disease. Affected individuals manifest failure to thrive, severe muscle weakness, spasticity and psychomotor retardation. Vision and hearing are impaired.
Polyol dehydrogenase that catalyzes the reversible NAD(+)-dependent oxidation of various sugar alcohols. Is mostly active with D-sorbitol (D-glucitol), L-threitol, xylitol and ribitol as substrates, leading to the C2-oxidized products D-fructose, L-erythrulose, D-xylulose, and D-ribulose, respectively (PubMed:3365415). Is a key enzyme in the polyol pathway that interconverts glucose and fructose via sorbitol, which constitutes an important alternate route for glucose metabolism. The polyol pathw
Mitochondrion membraneCell projection, cilium, flagellum
Neuronopathy, distal hereditary motor, autosomal recessive 8
An autosomal recessive disorder characterized by motor axonal neuropathy, slowly progressive distal muscle weakness mainly affecting the lower limbs, difficulty walking, and increased serum sorbitol. Additional variable features are distal sensory impairment, upper limb tremor, scoliosis, and mild hearing loss.
Acts as a guanine-nucleotide releasing factor (GEF) for Rab GTPases by promoting the conversion of inactive RAB-GDP to the active form RAB-GTP (PubMed:27103069, PubMed:27193190, PubMed:27617292, PubMed:28195531, PubMed:37821429). Acts as a GEF for RAB39A which enables HOPS-mediated autophagosome-lysosome membrane tethering and fusion in mammalian autophagy (PubMed:37821429). Component of the C9orf72-SMCR8 complex where both subunits display GEF activity and that regulates autophagy (PubMed:27103
CytoplasmNucleusCytoplasm, P-bodyCytoplasm, Stress granuleEndosomeLysosomeCytoplasmic vesicle, autophagosomeAutolysosomeSecretedCell projection, axonCell projection, growth conePerikaryonCell projection, dendritePresynapsePostsynapseNucleus membrane
Frontotemporal dementia and/or amyotrophic lateral sclerosis 1
An autosomal dominant neurodegenerative disorder characterized by adult onset of frontotemporal dementia and/or amyotrophic lateral sclerosis in an affected individual. There is high intrafamilial variation. Frontotemporal dementia is characterized by frontal and temporal lobe atrophy associated with neuronal loss, gliosis, and dementia. Patients exhibit progressive changes in social, behavioral, and/or language function. Amyotrophic lateral sclerosis is characterized by the death of motor neurons in the brain, brainstem, and spinal cord, resulting in fatal paralysis.
Transcription coactivator which associates with nuclear receptors, transcriptional coactivators including EP300, CREBBP and NCOA1, and basal transcription factors like TBP and TFIIA to facilitate nuclear receptors-mediated transcription (PubMed:10454579, PubMed:25219498). May thereby play an important role in establishing distinct coactivator complexes under different cellular conditions (PubMed:10454579, PubMed:25219498). Plays a role in thyroid hormone receptor and estrogen receptor transactiv
NucleusCytoplasm, cytosolCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Spinal muscular atrophy with congenital bone fractures 1
An autosomal recessive neuromuscular disorder characterized by prenatal-onset spinal muscular atrophy, multiple congenital contractures consistent with arthrogryposis multiplex congenita, respiratory distress, and congenital bone fractures.
Constitutes one of the two catalytic subunit of the tRNA-splicing endonuclease complex, a complex responsible for identification and cleavage of the splice sites in pre-tRNA. It cleaves pre-tRNA at the 5'- and 3'-splice sites to release the intron. The products are an intron and two tRNA half-molecules bearing 2',3'-cyclic phosphate and 5'-OH termini. There are no conserved sequences at the splice sites, but the intron is invariably located at the same site in the gene, placing the splice sites
NucleusNucleus, nucleolus
Pontocerebellar hypoplasia 2B
A disorder characterized by an abnormally small cerebellum and brainstem, and progressive microcephaly from birth combined with extrapyramidal dyskinesia. Severe chorea occurs and epilepsy is frequent. There are no signs of spinal cord anterior horn cells degeneration.
Fodrin, which seems to be involved in secretion, interacts with calmodulin in a calcium-dependent manner and is thus candidate for the calcium-dependent movement of the cytoskeleton at the membrane
Cytoplasm, cytoskeletonCytoplasm, cell cortex
Developmental and epileptic encephalopathy 5
A disorder characterized by seizures associated with hypsarrhythmia, profound intellectual disability with lack of visual attention and speech development, as well as spastic quadriplegia.
Involved in EGFR trafficking, acting as negative regulator of endocytic EGFR internalization at the plasma membrane
Cytoplasm
Spinocerebellar ataxia 2
Spinocerebellar ataxia is a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to cerebellum degeneration with variable involvement of the brainstem and spinal cord. SCA2 belongs to the autosomal dominant cerebellar ataxias type I (ADCA I) which are characterized by cerebellar ataxia in combination with additional clinical features like optic atrophy, ophthalmoplegia, bulbar and extrapyramidal signs, peripheral neuropathy and dementia. SCA2 is characterized by hyporeflexia, myoclonus and action tremor and dopamine-responsive parkinsonism. In some patients, SCA2 presents as pure familial parkinsonism without cerebellar signs.
The SMN complex catalyzes the assembly of small nuclear ribonucleoproteins (snRNPs), the building blocks of the spliceosome, and thereby plays an important role in the splicing of cellular pre-mRNAs (PubMed:18984161, PubMed:9845364). Most spliceosomal snRNPs contain a common set of Sm proteins SNRPB, SNRPD1, SNRPD2, SNRPD3, SNRPE, SNRPF and SNRPG that assemble in a heptameric protein ring on the Sm site of the small nuclear RNA to form the core snRNP (Sm core) (PubMed:18984161). In the cytosol,
Nucleus, gemNucleus, Cajal bodyCytoplasmCytoplasmic granulePerikaryonCell projection, neuron projectionCell projection, axonCytoplasm, myofibril, sarcomere, Z line
Spinal muscular atrophy 1
A form of spinal muscular atrophy, a group of neuromuscular disorder characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. Autosomal recessive forms are classified according to the age of onset, the maximum muscular activity achieved, and survivorship. The severity of the disease is mainly determined by the copy number of SMN2, a copy gene which predominantly produces exon 7-skipped transcripts and only low amount of full-length transcripts that encode for a protein identical to SMN1. Only about 4% of SMA patients bear one SMN1 copy with an intragenic mutation. SMA1 is a severe form, with onset before 6 months of age. SMA1 patients never achieve the ability to sit.
Plays a crucial role in the formation of lipid droplets (LDs) which are storage organelles at the center of lipid and energy homeostasis (PubMed:19278620, PubMed:21533227, PubMed:30293840, PubMed:31708432). In association with LDAF1, defines the sites of LD formation in the ER (PubMed:31708432). Also required for growth and maturation of small nascent LDs into larger mature LDs (PubMed:27564575). Mediates the formation and/or stabilization of endoplasmic reticulum-lipid droplets (ER-LD) contacts
Endoplasmic reticulum membraneLipid droplet
Lipodystrophy, congenital generalized, 2
A form of congenital generalized lipodystrophy, a metabolic disorder characterized by a near complete absence of adipose tissue, extreme insulin resistance, hypertriglyceridemia, hepatic steatosis and early onset of diabetes. Inheritance is autosomal recessive.
Converts O-phosphoseryl-tRNA(Sec) to selenocysteinyl-tRNA(Sec) required for selenoprotein biosynthesis
Cytoplasm
Pontocerebellar hypoplasia 2D
A disorder characterized by postnatal onset of progressive atrophy of the cerebrum and cerebellum, microcephaly, profound intellectual disability, spasticity, and variable seizures.
Non-selective calcium permeant cation channel involved in osmotic sensitivity and mechanosensitivity (PubMed:16293632, PubMed:18695040, PubMed:18826956, PubMed:22526352, PubMed:23136043, PubMed:29899501). Activation by exposure to hypotonicity within the physiological range exhibits an outward rectification (PubMed:18695040, PubMed:18826956, PubMed:29899501). Also activated by heat, low pH, citrate and phorbol esters (PubMed:16293632, PubMed:18695040, PubMed:18826956, PubMed:20037586, PubMed:219
Cell membraneApical cell membraneCell junction, adherens junctionCell projection, ciliumEndoplasmic reticulum
Brachyolmia 3
A form of brachyolmia, a clinically and genetically heterogeneous skeletal dysplasia primarily affecting the spine and characterized by a short trunk, short stature, and platyspondyly. BCYM3 is an autosomal dominant form with severe scoliosis with or without kyphosis, and flattened irregular cervical vertebrae.
Part of the dynactin complex that activates the molecular motor dynein for ultra-processive transport along microtubules (By similarity). Plays a key role in dynein-mediated retrograde transport of vesicles and organelles along microtubules by recruiting and tethering dynein to microtubules. Binds to both dynein and microtubules providing a link between specific cargos, microtubules and dynein. Essential for targeting dynein to microtubule plus ends, recruiting dynein to membranous cargos and en
CytoplasmCytoplasm, cytoskeletonCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centrioleCytoplasm, cytoskeleton, spindleNucleus envelopeCytoplasm, cell cortex
Neuronopathy, distal hereditary motor, autosomal dominant 14
A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs.
Involved in elastic and collagen fibers formation. It is required for EFEMP2 deposition into the extracellular matrix, and collagen network assembly and cross-linking via protein-lysine 6-oxidase/LOX activity (PubMed:36351433). May be responsible for anchoring smooth muscle cells to elastic fibers, and may be involved in the processes that regulate vessel assembly. Has cell adhesive capacity
Secreted, extracellular space, extracellular matrix
Neuronopathy, distal hereditary motor, autosomal dominant 10
A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular diseases caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. HMND10 is characterized by length-dependent motor neuropathy primarily affecting the lower limbs, and onset of distal muscle weakness and atrophy in early childhood resulting in walking difficulties and gait abnormalities. Some affected individuals have pyramidal signs, including hyperreflexia. More variable features may include mild intellectual disability, minor gyration defects on brain imaging, foot deformities, and connective tissue defects.
Necessary for the fragmentation of Golgi stacks during mitosis and for their reassembly after mitosis. Involved in the formation of the transitional endoplasmic reticulum (tER). The transfer of membranes from the endoplasmic reticulum to the Golgi apparatus occurs via 50-70 nm transition vesicles which derive from part-rough, part-smooth transitional elements of the endoplasmic reticulum (tER). Vesicle budding from the tER is an ATP-dependent process. The ternary complex containing UFD1, VCP and
Cytoplasm, cytosolEndoplasmic reticulumNucleusCytoplasm, Stress granule
Inclusion body myopathy with early-onset Paget disease with or without frontotemporal dementia 1
An autosomal dominant disease characterized by disabling muscle weakness clinically resembling to limb girdle muscular dystrophy, osteolytic bone lesions consistent with Paget disease, and premature frontotemporal dementia. Clinical features show incomplete penetrance.
Substrate recognition component of a SCF (SKP1-CUL1-F-box protein) E3 ubiquitin-protein ligase complex which mediates the ubiquitination and subsequent proteasomal degradation of target proteins (PubMed:20596027, PubMed:22632967, PubMed:26818844, PubMed:27080313, PubMed:27653696, PubMed:28852778). The SCF(CCNF) E3 ubiquitin-protein ligase complex is an integral component of the ubiquitin proteasome system (UPS) and links proteasome degradation to the cell cycle (PubMed:20596027, PubMed:26818844,
NucleusCytoplasm, perinuclear regionCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriole
Frontotemporal dementia and/or amyotrophic lateral sclerosis 5
A neurodegenerative disorder characterized by frontotemporal dementia and/or amyotrophic lateral sclerosis in affected individuals. There is high intrafamilial variation. Frontotemporal dementia is characterized by frontal and temporal lobe atrophy associated with neuronal loss, gliosis, and dementia. Patients exhibit progressive changes in social, behavioral, and/or language function. Amyotrophic lateral sclerosis is characterized by the death of motor neurons in the brain, brainstem, and spinal cord, resulting in fatal paralysis. FTDALS5 is an autosomal dominant form with age-dependent penetrance. Penetrance is estimated to be 50% by age 56 and 100% by age 61.
Non-catalytic component of the RNA exosome complex which has 3'->5' exoribonuclease activity and participates in a multitude of cellular RNA processing and degradation events. In the nucleus, the RNA exosome complex is involved in proper maturation of stable RNA species such as rRNA, snRNA and snoRNA, in the elimination of RNA processing by-products and non-coding 'pervasive' transcripts, such as antisense RNA species and promoter-upstream transcripts (PROMPTs), and of mRNAs with processing defe
CytoplasmNucleus, nucleolusNucleus
Pontocerebellar hypoplasia 1B
A severe autosomal recessive neurologic disorder characterized by a combination of cerebellar and spinal motor neuron degeneration beginning at birth. There is diffuse muscle weakness, progressive microcephaly, global developmental delay, and brainstem involvement.
Required for endoplasmic reticulum (ER) network formation, shaping and remodeling; it links ER tubules to the cytoskeleton. May also enhance the cell surface expression of odorant receptors (PubMed:20200447). May play a role in long-term axonal maintenance (PubMed:24478229)
MembraneMitochondrion membraneEndoplasmic reticulum
Spastic paraplegia 31, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Inhibits amyloid precursor protein processing, probably by blocking BACE1 activity (PubMed:15286784). Enhances trafficking of the glutamate transporter SLC1A1/EAAC1 from the endoplasmic reticulum to the cell surface (By similarity). Plays a role in the translocation of SLC2A4/GLUT4 from intracellular membranes to the cell membrane which facilitates the uptake of glucose into the cell (By similarity)
Endoplasmic reticulum membraneSarcoplasmic reticulum membraneCell membraneCell membrane, sarcolemmaCell membrane, sarcolemma, T-tubuleCytoplasm, myofibril, sarcomere, Z lineCytoplasm, cytoskeleton
Spastic paraplegia 12, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Acts as an adapter protein linking the dynein motor complex to various cargos and converts dynein from a non-processive to a highly processive motor in the presence of dynactin. Facilitates and stabilizes the interaction between dynein and dynactin and activates dynein processivity (the ability to move along a microtubule for a long distance without falling off the track) (PubMed:25814576). Facilitates the binding of RAB6A to the Golgi by stabilizing its GTP-bound form. Regulates coat complex co
Golgi apparatusCytoplasm, cytoskeletonCytoplasmNucleus envelopeNucleus, nuclear pore complex
Spinal muscular atrophy, lower extremity-predominant 2A, childhood onset, autosomal dominant
An autosomal dominant form of spinal muscular atrophy characterized by early-childhood onset of muscle weakness and atrophy predominantly affecting the proximal and distal muscles of the lower extremity, although some patients may show upper extremity involvement. The disorder results in delayed walking, waddling gait, difficulty walking, and loss of distal reflexes. Some patients may have foot deformities or hyperlordosis, and some show mild upper motor signs, such as spasticity. Sensation, bulbar function, and cognitive function are preserved. The disorder shows very slow progression throughout life.
5' to 3' helicase that unwinds RNA and DNA duplexes in an ATP-dependent reaction (PubMed:19158098, PubMed:22999958, PubMed:30218034). Specific to 5'-phosphorylated single-stranded guanine-rich sequences (PubMed:22999958, PubMed:8349627). May play a role in RNA metabolism, ribosome biogenesis or initiation of translation (PubMed:19158098, PubMed:19299493). May play a role in regulation of transcription (By similarity). Interacts with tRNA-Tyr (PubMed:19299493)
NucleusCytoplasmCell projection, axon
Neuronopathy, distal hereditary motor, autosomal recessive 1
A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs.
Catalyzes the attachment of tryptophan to tRNA(Trp) in a two-step reaction: tryptophan is first activated by ATP to form Trp-AMP and then transferred to the acceptor end of the tRNA(Trp) Has no angiostatic activity Possesses an angiostatic activity but has no aminoacylation activity (PubMed:11773625, PubMed:11773626, PubMed:14630953). Inhibits fluid shear stress-activated responses of endothelial cells (PubMed:14630953). Regulates ERK, Akt, and eNOS activation pathways that are associated with a
Cytoplasm
Neuronopathy, distal hereditary motor, autosomal dominant 9
A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMND9 is characterized by juvenile onset of slowly progressive distal muscle weakness and atrophy affecting both the lower and upper limbs.
May be involved in the maintenance of mitochondrial organization and mitochondrial cristae structure
Mitochondrion intermembrane space
Frontotemporal dementia and/or amyotrophic lateral sclerosis 2
A neurodegenerative disorder characterized by frontotemporal dementia and/or amyotrophic lateral sclerosis in affected individuals. There is high intrafamilial variation. Frontotemporal dementia is characterized by frontal and temporal lobe atrophy associated with neuronal loss, gliosis, and dementia. Patients exhibit progressive changes in social, behavioral, and/or language function. Amyotrophic lateral sclerosis is characterized by the death of motor neurons in the brain, brainstem, and spinal cord, resulting in fatal paralysis.
Promotes matrix assembly (By similarity). Involved in the organization of skeletal muscles and in the formation of neuromuscular junctions (Probable)
Secreted, extracellular space, extracellular matrix, basement membrane
Neuronopathy, hereditary motor, autosomal recessive 7
An autosomal recessive, neuromyopathic disorder that manifests in childhood or adulthood with proximal and distal muscle weakness predominantly of the lower limbs. Affected individuals have difficulty climbing stairs and problems standing on the heels. Most patients have foot deformities, and some may have leg muscle atrophy. Muscle biopsy and electrophysiologic studies are consistent with both a myopathic process and an axonal motor neuropathy.
High-affinity Na(+)-coupled choline transmembrane symporter (PubMed:11027560, PubMed:11068039, PubMed:12237312, PubMed:12969261, PubMed:17005849, PubMed:23132865, PubMed:23141292, PubMed:27569547). Functions as an electrogenic, voltage-dependent transporter with variable charge/choline stoichiometry (PubMed:17005849). Choline uptake and choline-induced current is also Cl(-)-dependent where Cl(-) is likely a regulatory ion rather than cotransported ion (PubMed:11068039, PubMed:12237312, PubMed:17
Presynaptic cell membraneCell projection, axonEarly endosome membraneCytoplasmic vesicle, secretory vesicle, synaptic vesicle membrane
Neuronopathy, distal hereditary motor, autosomal dominant 7
A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMND7 is characterized by onset in the second decade of progressive distal muscle wasting and weakness affecting the upper and lower limbs and resulting in walking difficulties and hand grip. There is significant muscle atrophy of the hands and lower limbs. The disorder is associated with vocal cord paresis due to involvement of the tenth cranial nerve.
Variantes genéticas (ClinVar)
310 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 64 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
180 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença das células dos cornos anteriores
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
3 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
1.295 ensaios clínicos encontrados, 4 ativos.
Publicações mais relevantes
Mostrando amostra de 200 publicações de um total de 3.353
Muscle MRI and Muscle Ultrasound Applications in MND/ALS: Academic Insights and Clinical Opportunities.
There is an unmet need for the clinically relevant ALS biomarkers to facilitate an accurate diagnosis in suspected cases, monitor disease progression and evaluate response to therapy in clinical trials. While the MND/ALS literature is dominated by innovative brain studies, motor disability in ALS is primarily driven by neurogenic muscle change impacting mobility, dexterity, respiratory and bulbar function. With the intention of raising awareness of muscle-derived imaging markers in ALS, a systematic review has been conducted. Study designs, imaging methods, data interpretation frameworks, and cohort characteristics were systematically evaluated to identify innovative approaches and barriers to clinical implementation. A total of 219 studies were screened and 73 original studies selected for systematic review; 37 muscle MRI studies and 36 studies using ultrasound, PET or CT. All of the selected studies successfully captured ALS-associated muscle degeneration and their methods included the evaluation of muscle dimensions (thickness/volumes n = 34), 'acute' denervation (water content, n = 15), fasciculation counts (n = 14), 'chronic' neurogenic change (fat content, n = 21), metabolic changes (n = 4), diffusion alterations (n = 8) and echo intensity changes (n = 13). Despite the huge impact of lower motor neuron dysfunction on the patients' independence, survival and quality of life, muscle imaging is a glaringly overlooked frontier of MND/ALS research. This is a missed opportunity, as a variety of non-invasive quantitative muscle imaging techniques have been successfully used in other neurological conditions; these protocols are easy to implement on commercial MRI and ultrasound platforms and recent studies have demonstrated their ease of use and potential clinical utility.
Historical and clinical analysis of a case of progressive muscular atrophy (1853-1871).
This article presents the historical and clinical analysis of Auguste-Joseph Bellinghen (1818-1872), whose medical course was documented in 1853 and later published in 1871. These records provide a rare longitudinal account of a patient with progressive muscular atrophy (PMA), a condition first described by Aran in 1850 and later redefined within the spectrum of motor neuron diseases by Charcot. The 1853 manuscript depicts a 35-year-old artisan with asymmetrical weakness, fasciculations-described by the patient as a "gelatinous oscillation"-and progressive muscular wasting, while the 1871 report shows him severely disabled, socially marginalized, and institutionalized. Together, these sources illustrate both the slow, relentless course of PMA and the therapeutic practices of the time, including cauterization, sulfur baths, and electrotherapy, all of which proved ineffective. The case also reflects the evolution of nosological frameworks: from Aran's clinical description without neuropathological confirmation to Charcot's integration of anatomical lesions into disease classification, thereby laying the foundations of modern neurology. Beyond clinical insights, the documents highlight the value of hospital archives, medical iconography, and the role of interns in constructing medical memory. Bellinghen's case thus exemplifies how individual trajectories illuminate both the biological and social dimensions of chronic illness in 19th-century France, while providing historiographic depth to the early history of motor neuron disease.
Paraspeckle condensation is controlled via TDP-43 polymerization and linked to neuroprotection.
The paraspeckle is a disease-relevant biomolecular condensate assembled from long non-coding RNA (lncRNA) NEAT1_2 ribonucleoprotein particles. Paraspeckle biogenesis is suppressed in normal tissues, yet it can be rapidly upregulated under stress. Here we demonstrate that a neurodegeneration-linked RNA-binding protein TDP-43 inhibits NEAT1_2 ribonucleoprotein particle condensation into the paraspeckle, in a concentration-dependent manner, which requires its intact polymerization and RNA binding. This effect is counterbalanced by core paraspeckle proteins such as FUS. Below disruptive concentrations, TDP-43 can be recruited into paraspeckles, forming non-liquid clusters. Under stress, TDP-43 sequestration into de novo nuclear condensates alleviates paraspeckle suppression and increases their dynamism. NEAT1_2 middle-part and 3'-end UG repeats mediate paraspeckle regulation by TDP-43 cotranscriptionally and post assembly, respectively. The deletion of the 3'-end UG repeat increases paraspeckle stability and cytoprotection in stressed human neurons. Consistently, longer 3'-end UG repeats are linked to shorter survival in the neurodegenerative disease amyotrophic lateral sclerosis. Thus, TDP-43 is a critical regulator of paraspeckle condensates linked to cytoprotection.
Identification of tofersen PD-response biomarkers in VALOR clinical trial CSF via multiplexed quantitative proteomics.
Tofersen, the first approved genetically targeted therapy for amyotrophic lateral sclerosis (ALS), demonstrates significant lowering of plasma neurofilament in adults carrying mutations in the superoxide dismutase 1 (SOD1) gene; however, additional biomarkers of treatment response in ALS are lacking. Here, we analyze longitudinally collected cerebrospinal fluid (CSF) samples from the phase 3 VALOR clinical trial to identify candidate tofersen treatment-response biomarkers in SOD1-ALS via quantitative proteomics. We observe significant modulation from baseline abundance for 56 proteins in tofersen-treated participants relative to placebo, including CSF GPNMB, which is significantly and continuously elevated across all post-baseline timepoints. We orthogonally confirm this observation by GPNMB immunoassay in independent tofersen-treated cohorts. Taken together, these data identify pharmacodynamic-response biomarkers of tofersen treatment that can be measured as early as 4 weeks post-treatment in SOD1-ALS patients and demonstrate the utility of leveraging unbiased proteomic screening integrated with targeted validation methods to identify pharmacodynamic-response biomarkers in clinical trial patient samples.
HML-2 env knockdown by AAV9-mediated miRNAs attenuates amyotrophic lateral sclerosis-like manifestations in mice.
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease for which there is no cure. While the precise etiology of ALS remains elusive, growing evidence suggests a pathogenic role for human endogenous retrovirus-K (HERV-K) in ALS. Expression of HERV-K subtype HML-2 envelope protein in neurons causes neurotoxicity in vitro and induces ALS-like symptoms in mice. We investigated the use of the Adeno-Associated Virus-9 (AAV9)-mediated artificial microRNA (amiRNA) targeting the HML-2 env gene in an ALS mouse model. From an in vitro screen of amiRNAs targeting the HML-2 env gene three were chosen and inserted in tandem into an AAV9 vector and validated in vitro. This approach provided robust silencing of the transgene, with tandem amiRNA achieving robust reduction in gene and protein expression levels. Its therapeutic effectiveness was tested in an HML-2 Env transgenic mouse model in which the env gene is expressed under the neuron-specific thy1 promoter and develops an ALS-like phenotype. A single intracerebroventricular injection of AAV9 vector encoding the amiRNAs into the mice at postnatal day 1 effectively reduced HML-2 Env expression in the brain and spinal cord at 84 days post-injection which was the longest time point studied. Knockdown of HML-2 env decreased the loss of cortical and spinal motor neurons and alleviated muscle fiber degeneration and fiber type grouping. This led to improved motor function. Our results provide compelling evidence supporting the use of multiple amiRNAs delivered in an AAV9 vector for treating forms of ALS linked to HML-2.
Publicações recentes
The effect of statins on the survival of patients with amyotrophic lateral sclerosis: a meta-analysis.
Challenges in medical care for amyotrophic lateral sclerosis: a survey of physicians from Republic of Bashkortostan (Russia), Belarus, and Kazakhstan.
The Impact on Systematic Reviews of Risk of Bias Assessment Changes From Conference Abstracts to Full Text.
Primary lateral sclerosis in Brazil: phenotypic heterogeneity, non-motor features, and prognostic markers in a 17-year multicentre cohort.
Between guidelines and reality: expert neurologists' perspectives on structural resources for ALS care in Germany.
📚 EuropePMC2.169 artigos no totalmostrando 199
Translation of the Dimensional Apathy Scale to Brazilian Portuguese to assess people living with HIV.
Dementia & neuropsychologiaWhy is it so difficult to discover drugs for amyotrophic lateral sclerosis? A protein intrinsic disorder perspective.
Expert opinion on drug discoveryThe role of N-acetylcysteine and glutathione in the management of Parkinson's disease: a systematic review of oxidative biomarkers and clinical outcomes.
Amino acidsMotor Neuron Disease Mortality Trends in Australia From 1986 to 2023: A Population-Based Study.
The Medical journal of AustraliaMuscle MRI and Muscle Ultrasound Applications in MND/ALS: Academic Insights and Clinical Opportunities.
European journal of neurologyHirayama disease: an uncommon cause of motor neuron disease.
Arquivos de neuro-psiquiatriaHistorical and clinical analysis of a case of progressive muscular atrophy (1853-1871).
European neurologyParaspeckle condensation is controlled via TDP-43 polymerization and linked to neuroprotection.
Nature cell biologyMultisystemic Involvement in Autosomal Recessive Cerebellar Ataxia Type 8 Having a Novel SYNE1 Nonsense Variant.
Internal medicine (Tokyo, Japan)Identification of tofersen PD-response biomarkers in VALOR clinical trial CSF via multiplexed quantitative proteomics.
Cell reports. MedicineTreating age-related loss of muscle mass and function: Where should we be focusing?
The Journal of physiologyHML-2 env knockdown by AAV9-mediated miRNAs attenuates amyotrophic lateral sclerosis-like manifestations in mice.
Brain : a journal of neurologyMotor Neuron Disease with Guillain-Barré Syndrome? Motor Band Sign with Anti-GQ1b Antibodies.
Diagnostics (Basel, Switzerland)Impaired Acetyl-CoA Compartmentalization Drives a Futile Lipogenic-Oxidative Cycle in N88S Seipinopathy.
CellsImmune imbalance between T helper 1, T helper 17 and regulatory T cells fuels amyotrophic lateral sclerosis pathogenesis: disease trajectory, diagnosis and therapeutic implications.
Journal of neuroinflammationMotor Neuronopathy With Widespread Fasciculations in MCM3AP-Related Disorder: Clinical and Muscle MRI Insights.
Journal of the peripheral nervous system : JPNSCIDP With and Without Monoclonal Gammopathy of Undetermined Significance (MGUS): Comparison of Clinical Phenotype, Diagnostic Features, and Treatment Response.
Journal of the peripheral nervous system : JPNSOral Health in Amyotrophic Lateral Sclerosis: Feasibility of Oral Screening and Determinants of Poor Outcomes.
Muscle & nerveWhy we need to depict the structure of DNA correctly: it's so groovy.
Brain : a journal of neurologyInnovation, Adaptation, and Human Dignity in Assistive Robotics in Amyotrophic Lateral Sclerosis: A Rehabilitation Medicine Perspective.
Journal of biotechnology and biomedicineMotor neuron disease can present as a paraneoplastic neurologic syndrome with various phenotypes.
Brain communicationsALS untangled #83: clenbuterol.
Amyotrophic lateral sclerosis & frontotemporal degenerationExpanding the Motor Band Sign in Motor Neuron Disease Using 7T MRI: Visualization of Cortical Layer-Dependent Iron Deposition in the Primary Motor Cortex.
Muscle & nerveBDNF relates to prefrontal cortex activity in the context of physical exercise.
Brain research"What about me? I'm supposed to be … superhuman?": exploring staff perspectives on how to deliver high quality psychological care for people living with amyotrophic lateral sclerosis.
Neurodegenerative disease managementMapping end-of-life care for patients with neurological conditions in German hospices: a point prevalence survey.
BMJ neurology openGadolinium enhancement of the cauda equina in a case of familial ALS with p.S135G SOD1 mutation.
Amyotrophic lateral sclerosis & frontotemporal degenerationExploring the use of narrative-based approaches in individuals with amyotrophic lateral sclerosis: A narrative review.
Palliative & supportive careProspective Validation of the New PLS Diagnostic Criteria From PLS Natural History Study: EMG and Neurofilament Analyses.
Muscle & nerveRespiratory Onset Amyotrophic Lateral Sclerosis in a Patient With C9orf72 Expansion.
Journal of clinical neuromuscular diseaseVoltage-gating and neuronal signalling in neurodegeneration: From neuropathology to therapeutic opportunities in motor neuron disease.
Neurobiology of diseaseKIF5A and ALS: a clinical and genetic description of a case series and review of literature.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyImaging spectrum and diagnostic insights in Hirayama disease: A case series.
Radiology case reportsMaximal motor unit firing rates decline with amyotrophic lateral sclerosis progression.
Clinical neurophysiology : official journal of the International Federation of Clinical NeurophysiologyAnalysis and comparison of the trends in the burden of motor neuron disease in China and worldwide from 1990 to 2021.
PloS oneBlood Lactate as a Prognostic Biomarker for Survival and Weight Loss in Amyotrophic Lateral Sclerosis: An Exploratory-Validation Study.
Annals of neurologyExploring Patient Perspectives on an Immersive Virtual Reality Experience for Neuromuscular Diseases.
Games for health journalDisruption of BDNF signalling in neuropathologies.
Biochemical Society transactionsStanding up to neurodegeneration: evaluating autonomic stress and safety in sit-to-stand using heart rate variability analysis.
Biomedizinische Technik. Biomedical engineeringImaging-derived neuromuscular ultrasound phenotypes are associated with functional status in amyotrophic lateral sclerosis.
Journal of neurologyAccuracy of clinical diagnosis in neurodegenerative diseases - a study of 455 autopsy cases.
Journal of neurologySkeletal muscle in spinal muscular atrophy: Critical insights from pathogenesis to therapeutic strategies.
Neurobiology of diseaseThe Role of the Golgi Apparatus in Neurodegeneration.
Sub-cellular biochemistryTiming of communication and technology control support in ALS - a systematic review.
Amyotrophic lateral sclerosis & frontotemporal degenerationPredictive genetic testing in amyotrophic lateral sclerosis (ALS): Experiences of decision-making and engagement with UK genetic counseling services.
Journal of genetic counselingDifferential inclusion formation of an aggregation-prone protein reveals differences in the proteostasis capacity of neuronal cell lines.
Biochimica et biophysica acta. Molecular cell researchAn implementation study of a remote monitoring platform to increase access to specialist care in motor neuron disease.
Neurodegenerative disease managementEfficient induction of motor neuron disease in transgenic G93A SOD1 mice by prion-like seeding.
PrionSynaptic and cytoskeletal CSF signatures of motor neuron disease: the role of cyclase-associated protein 2.
Scientific reportsSystemic immune-metabolic interactions in patients with amyotrophic lateral sclerosis: correlations of plasma cytokines with metabolic status.
Amyotrophic lateral sclerosis & frontotemporal degenerationExtracellular vesicles at the neuromuscular junction: messengers of synaptic health and disease.
Cell and tissue researchNatural Killer Cell Dysregulation During ALS Disease Progression: A Gene Expression Analysis.
Neurology open accessCan patient-reported outcome measures predict mortality in neurological populations? A systematic review.
Frontiers in neurologyMyelin sheaths in the central nervous system can withstand damage and dynamically remodel.
Science (New York, N.Y.)From Dish to Trial: Building Translational Models of ALS.
CellsFour decades of ALS care: a retrospective study of epidemiology, clinical course and changes in management.
Amyotrophic lateral sclerosis & frontotemporal degenerationImplications of virus-induced stress granules in tauopathies.
Translational neurodegenerationAnatomical reduction for focal fasciculations in peroneus brevis spondylolisthesis: a case report suggesting a mechanism of peripherally derived tremor.
BMC musculoskeletal disordersLost in translation: absence of KIAA1324/ELAPOR1 protein in pathological TDP-43-affected neurons in ALS/FTD.
Acta neuropathologica communicationsImpaired BDNF-TrkB trafficking and signalling in Down syndrome basal forebrain neurons.
Cell death & disease[Combination of amyotrophic lateral sclerosis with Alzheimer's disease].
Zhurnal nevrologii i psikhiatrii imeni S.S. KorsakovaCase Report: Spinal muscular atrophy with IgA nephropathy: a coincidence or association?
Frontiers in pediatricsA Systematic Review of Hyperselective Neurectomy for Management of the Spastic Upper Limb.
Hand (New York, N.Y.)Non-Cell-Autonomous Mechanisms and Systemic Interactions in Spinal Muscular Atrophy.
The American journal of pathologyChanges in body composition and phase angle from diagnosis to gastrostomy, in motor neuron disease patients: a longitudinal study.
European journal of clinical nutritionTrajectory of Mobility Function Decline for People With Motor Neuron Disease.
Archives of physical medicine and rehabilitationCurrent and Ongoing Clinical Studies.
Muscle & nerveAge-period-cohort effect on motor neuron disease mortality in the United States, 2001-2020.
Frontiers in neurologyTBK1-Associated Primary Lateral Sclerosis Followed by Right Temporal Variant Frontotemporal Dementia.
Annals of clinical and translational neurologyA multi-omics study on monozygotic twins discordant for amyotrophic lateral sclerosis and literature review underline a potential role for innate immunity and epigenetic dysregulation in disease mechanisms.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyTBK1 activity regulates the directionality of axonal transport of signalling endosomes.
Life science allianceElectrodiagnostic Approach to Defects of Neuromuscular Transmission.
Muscle & nerveCrossroad of motor neuron disease and dementia: Insights from TDP-43 RNA-binding deficiency.
Neural regeneration researchNational and subnational burden of neurological disorders in Iran, 1990 to 2021: results from the global burden of disease study 2021.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyA national survey of pseudobulbar affect and symptomatic treatment in Amyotrophic Lateral Sclerosis.
Amyotrophic lateral sclerosis & frontotemporal degenerationPrevalence and Clinical Associations of A-Waves in Routine Nerve Conduction Studies: A Retrospective Analysis.
Noro psikiyatri arsiviThe spinal and bulbar muscular atrophy-health index: a disease-specific outcome measure.
Amyotrophic lateral sclerosis & frontotemporal degenerationMolecular Characterization of Calu-3 Cells from Submerged to Air-Liquid Interface to Model Lung Infections.
Journal of proteome researchPleiotrophin/Midkine Pathway Is Dysregulated in a TDP-43A315T Mouse Model of Amyotrophic Lateral Sclerosis (ALS).
Neuropathology : official journal of the Japanese Society of NeuropathologyABCA1 acts as a protective modulator in amyotrophic lateral sclerosis.
iScienceThe genetics of autosomal recessive ALS: a review of the common forms and their phenotypes.
Amyotrophic lateral sclerosis & frontotemporal degenerationTargeting Non-coding RNAs in Neurodegeneration: Advances in Therapeutic RNA Modalities and Next-Gen Delivery Technologies.
Current Alzheimer researchExploring rare coding variants in UK biobank: preliminary associations with motor neuron disease.
Frontiers in aging neurosciencePain and fatigue in amyotrophic lateral sclerosis: a multiple methods study.
Neurodegenerative disease managementReduced osteogenic factors and early osteoblast senescence in SOD1(G93A) ALS mouse model.
JCI insightLiver Steatosis in Induced Hepatocytes From Carriers of Spinal Muscular Atrophy.
Muscle & nerveAddendum to the 2023 clinical practice guidelines for amyotrophic lateral sclerosis in Japan: approval and integration of novel disease-modifying therapies.
Rinsho shinkeigaku = Clinical neurologyDevelopment and validation of predictive models for 6-month gastrostomy timing in amyotrophic lateral sclerosis.
BMJ neurology openLong-Term Exposure to Air Pollution and Risk and Prognosis of Motor Neuron Disease.
JAMA neurologyAir Pollution and Motor Neuron Disease-Particulate-ly Risky.
JAMA neurologyExpanding the neurological spectrum of HTLV-1 beyond HAM/TSP: a contemporary perspective.
Lancet regional health. AmericasLAMP1 and LAMP2A localise to axonal organelles with distinct motility dynamics and partially overlapping molecular signatures in human neurons.
Journal of cell scienceWDR62 is required for proper proliferation and early differentiation of skeletal myoblasts.
Communications biologyEstablishing Diagnostic and Differential Diagnostic Criteria for Amyotrophic Lateral Sclerosis.
Journal of clinical medicineHeterogeneous phenotype and cardiovascular comorbidities in Swedish patients with spinobulbar muscular atrophy.
Journal of neurologyDemographic, clinical and genetic characteristics of patients with amyotrophic lateral sclerosis from two specialised centres in Austria.
Journal of neurologyNeuroinvasive West Nile Virus Presenting as Subacute Progressive Quadriparesis and Intractable Pain: A Case Report.
Case reports in neurological medicineFrontotemporal dementia: Clinical aspects, genetics, and neuropathology of a family with a C9ORF72 expansion in Argentina.
Brain pathology (Zurich, Switzerland)PBX-dependent and independent Hox programs establish and maintain motor neuron terminal Identity.
bioRxiv : the preprint server for biologyLysosomal escape and TMEM106B fibrillar core determine TDP-43 seeding outcomes.
bioRxiv : the preprint server for biologyMicroglia in C9orf72-associated amyotrophic lateral sclerosis: More or less active?
Neural regeneration researchPrevalence and trajectories of post-COVID-19 neuromuscular conditions: A systematic-review and meta-analysis.
Journal of the neurological sciencesDropped head syndrome leading to the diagnosis of antisynthetase syndrome: a motor neuron mimicker.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyInvestigating the pathogenic role of calpain proteases and the therapeutic potential of their inhibition in mice modelling Machado-Joseph disease.
Human molecular geneticsPrioritizing neuropsychological research and care in Amyotrophic Lateral Sclerosis (ALS): building an international neuropsychological framework for ALS.
Amyotrophic lateral sclerosis & frontotemporal degenerationFoot that spoke first: microvascular signs of a motor neuron disease.
Archives of disease in childhoodMicroglial HVCN1 Deficiency Improves Movement and Survival of SOD1G93A ALS Mice by Enhancing Microglial Migration and Neuroprotection.
Advanced science (Weinheim, Baden-Wurttemberg, Germany)Co-aggregation of annexin A11 and TDP-43 in FTLD/MND with primary lateral sclerosis phenotype.
Acta neuropathologica communicationsAAV9 gene therapy optimization for SMARD1/CMT2S: safety and long-term efficacy comparison of two vectors in a SMARD1 preclinical model.
Journal of biomedical scienceHereditary transthyretin amyloidosis with hand weakness and bulbar involvement.
Practical neurologyHuman Mutant Dynactin Subunit 1 Causes Profound Motor Neuron Disease Consistent with Possible Mechanisms Involving Axonopathy, Mitochondriopathy, Protein Nitration, and T-Cell-Mediated Cytolysis.
BiomoleculesMotor band sign in 18F-FDG PET/CT studies: a biomarker of degenerative upper motor neuron disease? A study of three cases and literature review.
NeurologiaMAPT p.V363I mutation in a patient with presenile dementia and late amyotrophic lateral sclerosis.
Amyotrophic lateral sclerosis & frontotemporal degenerationBody Weight and Range of Motion as Predictors of Trunk Asymmetry in Children With Spinal Muscular Atrophy: A Prospective Functional Assessment.
Medical science monitor : international medical journal of experimental and clinical researchSOD1 mutations in Taiwanese ALS patients: Clinical characteristics, frequency, and a p.T138R founder effect.
Amyotrophic lateral sclerosis & frontotemporal degenerationIncidence of MND in Canterbury, New Zealand and impact of a key worker role on hospital admissions.
Amyotrophic lateral sclerosis & frontotemporal degenerationDYNC1H1 in Spinal Muscular Atrophy: Diagnostic Findings From Two Families and a Comprehensive Review of Its Role in Neuromuscular and Neurodevelopmental Disorders.
Molecular genetics & genomic medicineSynaptic changes contribute to persistent extra-motor behaviour deficits in amyotrophic lateral sclerosis.
Acta neuropathologica communicationsPredicting Phenoconversion to Clinically Manifest ALS: Results of a Large-Scale Proteomic Study.
medRxiv : the preprint server for health sciencesHippocampal Subfield Integrity and Age-Driven Neural Correlates of Appetite Loss in Amyotrophic Lateral Sclerosis.
Journal of magnetic resonance imaging : JMRI10H-phenothiazine exerts beneficial effects in spinal muscular atrophy in vitro and in vivo models.
Scientific reportsA 10-year service evaluation of a multidisciplinary neuro-palliative care model in motor neuron disease: Impact on palliative care service delivery & advance care planning.
Palliative medicineAge-dependent removal of Atg9-containing vesicle accumulations in motoneuron disease models by physical exercise.
Translational neurodegenerationRole of Neuronal Cholecystokinin Receptor: An Emerging Therapeutic Target for Ameliorating Neurological Diseases.
Molecular neurobiologyClinical relevance of zebrafish for gene variants testing. Proof-of-principle with SMN1/SMA.
EMBO molecular medicineBiomarkers in ALS trials: from discovery to clinical utility.
Frontiers in neuroscienceU7 small nuclear RNA splice-switching therapeutics for STMN2 and UNC13A in Amyotrophic Lateral Sclerosis.
bioRxiv : the preprint server for biologyCell type-specific functions of the PBAF chromatin-remodeling complex in neuronal diversification.
Nature communicationsProtocol for applying expansion microscopy to the study of mammalian neuromuscular junctions.
STAR protocolsTargeted BDNF upregulation via upstream open reading frame disruption.
Molecular therapy : the journal of the American Society of Gene TherapyMulti-omics analyses identify potential epigenetic loci associated with survival in amyotrophic lateral sclerosis across diverse populations.
EBioMedicineHeteroaggregation of Wild-Type and ALS Mutant SOD1.
ACS chemical neuroscienceTrends in motor neuron disease mortality in the United States from 1999 to 2020.
Neurodegenerative disease managementExtracellular Matrix Remodeling in Motor Neuron Diseases.
International journal of molecular sciencesDigital App for Speech and Health Monitoring Study (DASH): protocol for a prospective longitudinal case-control observational study for developing speech datasets in neurodegenerative disorders and dementia.
BMJ openEarly assessment in bulbar-onset amyotrophic lateral sclerosis detects similar rates of nocturnal desaturation and orthopnoea compared to non-bulbar-onset disease.
Sleep & breathing = Schlaf & AtmungInsights and considerations on predicting cognitive and behavioral disturbances in MND with pure motor onset.
Journal of the neurological sciencesCase Series Assessing the Use of Levetiracetam for Gait Improvement in Primary Lateral Sclerosis.
The Canadian journal of neurological sciences. Le journal canadien des sciences neurologiquesHuman TDP-43 overexpression in zebrafish motor neurons triggers MND-like phenotypes through gain-of-function mechanism.
Acta neuropathologica communicationsMovement Related Beta-Band Modulation with OPM-MEG: A Pilot Study.
Brain topographySYNE1 Deficiency Manifesting Primarily With Motor Neuron Disease.
Neurology. GeneticsEmergent technologies and applications of TMS and TMS-EEG in clinical neurophysiology for early and differential diagnosis: IFCN handbook chapter.
Clinical neurophysiology : official journal of the International Federation of Clinical NeurophysiologyStartle Reflex in Primary Lateral Sclerosis (PLS): A Comparison With Amyotrophic Lateral Sclerosis (ALS).
Muscle & nerveThe Epidemiology of Primary Lateral Sclerosis: Results from a Population-Based Cohort.
Annals of neurologyGlobal vs. segmental acoustic features for dysarthria assessment in motor neuron diseases.
Computers in biology and medicinePhysical Therapist Interventions for People With Amyotrophic Lateral Sclerosis Across Disease Stages: A Systematic Review of Efficacy.
Physical therapySLP2/PHB Aggregates in ALS Mouse Models and Patients: Implications Beyond CHCHD10-Associated Motor Neuron Disease.
International journal of molecular sciencesAmyotrophic Lateral Sclerosis (ALS)-Related Mortality Among World Trade Center-Exposed and Non-World Trade Center-Exposed Rescue and Recovery Workers.
International journal of environmental research and public healthVenom Peptides Across Asian and American Tarantulas Utilize Dual Pharmacology to Target Activation and Fast Inactivation of Voltage-Gated Sodium Channels.
ToxinsMills' syndrome and myasthenia gravis: a case report.
Neuromuscular disorders : NMDLarge-scale drug screening in iPSC-derived motor neurons from sporadic ALS patients identifies a potential combinatorial therapy.
Nature neuroscienceAmyotrophic lateral sclerosis in Saudi Arabia: a multicenter descriptive study.
Amyotrophic lateral sclerosis & frontotemporal degenerationIsomiR utility in amyotrophic lateral sclerosis prognostication.
Med (New York, N.Y.)The genetic architecture of primary lateral sclerosis in a cohort of Italian patients.
Journal of neurologyTofacitinib extends survival in a mouse model of ALS through NK cell-independent mechanisms.
Frontiers in immunologyAnalytical and clinical validation of step counting method in people living with amyotrophic lateral sclerosis.
Scientific reportsApathy self-awareness and its neural correlates in Parkinson's Disease.
NPJ Parkinson's diseaseSynaptopathy in the TDP-43ΔNLS Mouse Model of Sporadic Amyotrophic Lateral Sclerosis.
The European journal of neuroscienceRole of 2-[18F]FDG-PET as a biomarker of upper motor neuron involvement in amyotrophic lateral sclerosis.
Journal of neurologyMifepristone alone and in combination with scAAV9-SMN1 gene therapy improves disease phenotypes in Smn2B/- spinal muscular atrophy mice.
Scientific reportsAn Autopsy Case of ALS Which Clinically Presented Sporadic Adult-Onset Lower Motor Neuron Disease and Genetically Had p. Leu127Ser (L126S) Variant in SOD1 and SMN2 Deletion.
Neuropathology : official journal of the Japanese Society of NeuropathologyValidation of the dimensional apathy scale and predictors of apathy in stroke survivors.
Scientific reportsThe structure, redox chemistry and motor neuron toxicity of heterodimeric zinc-deficient SOD1-implications for the toxic gain of function observed in ALS.
Neurobiology of diseaseExpanding the spectrum of annexin A11 proteinopathy in frontotemporal lobar degeneration and motor neuron disease.
Acta neuropathologicaSpinocerebellar Ataxia Type 3 Accompanied by Amyotrophic Lateral Sclerosis: A Case Report and Comprehensive Literature Review.
Internal medicine (Tokyo, Japan)The global burden of motor neuron disease: a systematic and additional analysis of global burden disease study 2021.
Orphanet journal of rare diseasesProtocol for in vivo assessment of glucose metabolism in mouse models.
STAR protocolsFacial onset sensory and motor neuronopathy: a diagnostic challenge.
Acta neurologica BelgicaCRISPR/Cas9 gene editing in muscle-related genetic disorders: Restoring function and exercise capacity.
Tissue & cellEarly brain-wide disruption of sleep microarchitecture in amyotrophic lateral sclerosis.
The Journal of clinical investigationUnraveling the genetic landscape of ALS in Greece: identification of known and novel causative variants in a 353-patient cohort.
Amyotrophic lateral sclerosis & frontotemporal degenerationALSUntangled #81: Pyridostigmine (mestinon®).
Amyotrophic lateral sclerosis & frontotemporal degenerationPrevalence of living alone with dementia and other progressive neurological conditions: findings from primary care data in England.
BMC medicineDTI changes of brachial plexus nerve roots in amyotrophic lateral sclerosis and their correlation with electrophysiology.
European radiology experimentalEfficacy and safety of risdiplam in adults with 5q-associated spinal muscular atrophy: a nationwide observational cohort study in Austria.
EClinicalMedicineRYR 1 Gene Mutation in Motor Neuron Disease: A 10-Year Case Observation.
Case reports in neurological medicineRehabilitation in Neuromuscular Diseases: Best Turkish Practice Recommendations by Multidisciplinary Experts.
Acta neurologica BelgicaExamining the impact and role of lipid classes on the risk of amyotrophic lateral sclerosis (ALS) onset: a systematic review and GRADE analysis of the evidence.
Amyotrophic lateral sclerosis & frontotemporal degenerationTDP-43-dependent mis-splicing of KCNQ2 triggers intrinsic neuronal hyperexcitability in ALS/FTD.
Nature neuroscienceEdaravone for amyotrophic lateral sclerosis.
Australian prescriberClinical Reasoning and Diagnostic Challenge in a 23-Year-Old Man With Rapidly Progressive Dysphagia and Hypophonia: Juvenile-Onset Amyotrophic Lateral Sclerosis Caused by a FUS Gene Mutation.
CureusDynamometric measurement of hand grip and pinch strength as functional independency outcome in neuromuscular diseases.
European journal of physical and rehabilitation medicineThe Other Side of the Same Coin: Beyond the Coding Region in Amyotrophic Lateral Sclerosis.
Pharmaceuticals (Basel, Switzerland)Emerging Approaches to Mitigate Neural Cell Degeneration with Nanoparticles-Enhanced Polyelectrolyte Systems.
MembranesAcceptance and Commitment Therapy for people living with motor neuron disease: the COMMEND feasibility study and randomised controlled trial.
Health technology assessment (Winchester, England)Hirayama disease: A case report.
Radiology case reportsStructure and Molecular Basis for Inhibiting Human SOD1 Aggregation by a Promising Decan Derivative Modulator: A Potential Therapeutic Strategy for Treating Amyotrophic Lateral Sclerosis (ALS).
ACS chemical neuroscienceSelf-reported initiation apathy is related to worse quality of life in amyotrophic lateral sclerosis.
Amyotrophic lateral sclerosis & frontotemporal degenerationTDP-43 loss induces cryptic polyadenylation in ALS/FTD.
Nature neuroscienceTDP-43 nuclear loss in FTD/ALS causes widespread alternative polyadenylation changes.
Nature neuroscienceUnraveling the Military Service-MND Connection: Time Frame, Exposures, and Phenotypic Considerations.
European journal of neurology[A case of adult-onset Krabbe disease diagnosed by galactocerebrosidase gene mutations, presenting with an atypical phenotype].
Rinsho shinkeigaku = Clinical neurologyRethinking ALS: Current understanding and emerging therapeutic strategies.
AIMS neuroscienceConditional ATXN2L-Null in Adult Frontal Cortex CamK2a+ Neurons Does Not Cause Cell Death but Restricts Spontaneous Mobility and Affects the Alternative Splicing Pathway.
CellsIs TREM2 a Stretch: Implications of TREM2 Along Spinal Cord Circuits in Health, Aging, Injury, and Disease.
CellsIntegrative treatment of the motor neuron disease amyotrophic lateral sclerosis, efficacy of pharmacotherapy, traditional Chinese medicine and importance of respiratory support, life-style, and gastrostomy-assisted nutrition: A review.
International journal of clinical pharmacology and therapeuticsUnraveling the Potential of Stem Cell Therapy in Motor Neuron Disease: A Narrative Review.
CNS & neurological disorders drug targetsBeyond motor neurons: autonomic dysfunction and ECG findings in adults with 5q-spinal muscular atrophy.
Journal of neurology[Clinical analysis of a motor neuron disease-like phenotype associated with anti-IgLON5 disease].
Zhonghua nei ke za zhiAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença das células dos cornos anteriores.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença das células dos cornos anteriores
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Muscle MRI and Muscle Ultrasound Applications in MND/ALS: Academic Insights and Clinical Opportunities.
- Historical and clinical analysis of a case of progressive muscular atrophy (1853-1871).
- Paraspeckle condensation is controlled via TDP-43 polymerization and linked to neuroprotection.
- Identification of tofersen PD-response biomarkers in VALOR clinical trial CSF via multiplexed quantitative proteomics.
- HML-2 env knockdown by AAV9-mediated miRNAs attenuates amyotrophic lateral sclerosis-like manifestations in mice.
- The effect of statins on the survival of patients with amyotrophic lateral sclerosis: a meta-analysis.
- Challenges in medical care for amyotrophic lateral sclerosis: a survey of physicians from Republic of Bashkortostan (Russia), Belarus, and Kazakhstan.
- The Impact on Systematic Reviews of Risk of Bias Assessment Changes From Conference Abstracts to Full Text.
- Primary lateral sclerosis in Brazil: phenotypic heterogeneity, non-motor features, and prognostic markers in a 17-year multicentre cohort.
- Between guidelines and reality: expert neurologists' perspectives on structural resources for ALS care in Germany.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:98503(Orphanet)
- MONDO:0020128(MONDO)
- GARD:19477(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q3221083(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
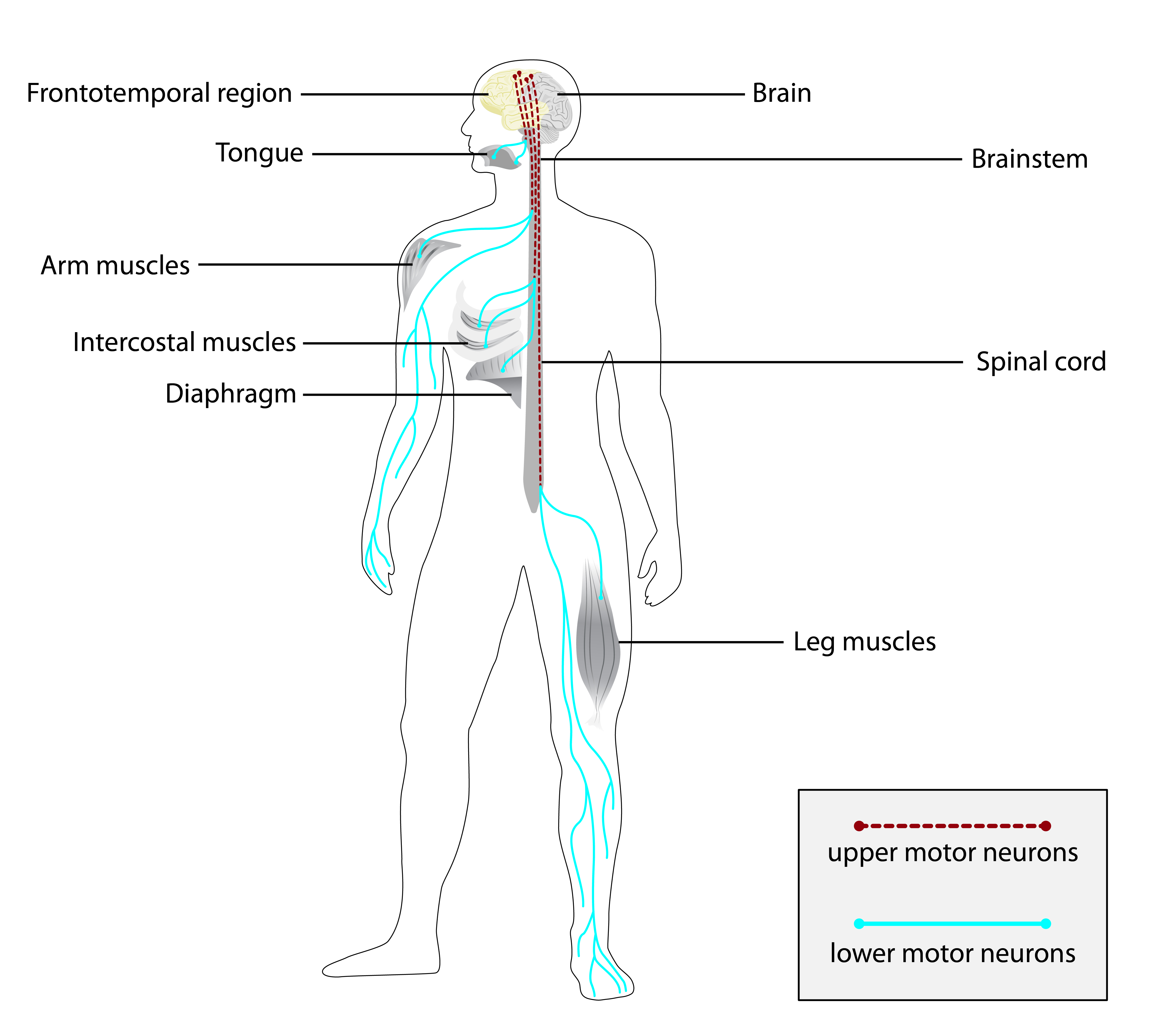
Doença das células dos cornos anteriores
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov