A hipoplasia pontocerebelar tipo 2 (PCH2) é o subtipo mais comum de hipoplasia pontocerebelar, caracterizada por início neonatal e falta de desenvolvimento motor voluntário e posteriormente microencefalia progressiva, clônus generalizado, desenvolvimento de coreia e espasticidade. A maioria dos pacientes não atingirá a puberdade.
Introdução
O que você precisa saber de cara
A hipoplasia pontocerebelar tipo 2 (PCH2) é o subtipo mais comum de hipoplasia pontocerebelar, caracterizada por início neonatal e falta de desenvolvimento motor voluntário e posteriormente microencefalia progressiva, clônus generalizado, desenvolvimento de coreia e espasticidade. A maioria dos pacientes não atingirá a puberdade.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 30 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 87 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
5 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Converts O-phosphoseryl-tRNA(Sec) to selenocysteinyl-tRNA(Sec) required for selenoprotein biosynthesis
Cytoplasm
Pontocerebellar hypoplasia 2D
A disorder characterized by postnatal onset of progressive atrophy of the cerebrum and cerebellum, microcephaly, profound intellectual disability, spasticity, and variable seizures.
Constitutes one of the two catalytic subunit of the tRNA-splicing endonuclease complex, a complex responsible for identification and cleavage of the splice sites in pre-tRNA. It cleaves pre-tRNA at the 5'- and 3'-splice sites to release the intron. The products are an intron and two tRNA half-molecules bearing 2',3'-cyclic phosphate and 5'-OH termini. There are no conserved sequences at the splice sites, but the intron is invariably located at the same site in the gene, placing the splice sites
NucleusNucleus, nucleolus
Pontocerebellar hypoplasia 2B
A disorder characterized by an abnormally small cerebellum and brainstem, and progressive microcephaly from birth combined with extrapyramidal dyskinesia. Severe chorea occurs and epilepsy is frequent. There are no signs of spinal cord anterior horn cells degeneration.
Non-catalytic subunit of the tRNA-splicing endonuclease complex, a complex responsible for identification and cleavage of the splice sites in pre-tRNA. It cleaves pre-tRNA at the 5' and 3' splice sites to release the intron. The products are an intron and two tRNA half-molecules bearing 2',3' cyclic phosphate and 5'-OH termini (PubMed:15109492, PubMed:27392077). There are no conserved sequences at the splice sites, but the intron is invariably located at the same site in the gene, placing the sp
NucleusNucleus, nucleolus
Pontocerebellar hypoplasia 2F
A neurodevelopmental disorder characterized by progressive microcephaly, cognitive and motor delay, poor or absent speech, seizures, and spasticity. PCH2F inheritance is autosomal recessive.
Non-catalytic subunit of the tRNA-splicing endonuclease complex, a complex responsible for identification and cleavage of the splice sites in pre-tRNA. It cleaves pre-tRNA at the 5' and 3' splice sites to release the intron. The products are an intron and two tRNA half-molecules bearing 2',3' cyclic phosphate and 5'-OH termini. There are no conserved sequences at the splice sites, but the intron is invariably located at the same site in the gene, placing the splice sites an invariant distance fr
NucleusNucleus, nucleolus
Pontocerebellar hypoplasia 4
A disorder characterized by an abnormally small cerebellum and brainstem, severe neonatal encephalopathy, microcephaly, myoclonus and muscular hypertonia. There is a severe inferior olivary and pontine neuronal loss and a diffuse white matter gliosis.
Constitutes one of the two catalytic subunit of the tRNA-splicing endonuclease complex, a complex responsible for identification and cleavage of the splice sites in pre-tRNA. It cleaves pre-tRNA at the 5'- and 3'-splice sites to release the intron. The products are an intron and two tRNA half-molecules bearing 2',3'-cyclic phosphate and 5'-OH termini. There are no conserved sequences at the splice sites, but the intron is invariably located at the same site in the gene, placing the splice sites
NucleusNucleus, nucleolus
Pontocerebellar hypoplasia 2C
A disorder characterized by an abnormally small cerebellum and brainstem, and progressive microcephaly from birth combined with extrapyramidal dyskinesia. Severe chorea occurs and epilepsy is frequent. There are no signs of spinal cord anterior horn cells degeneration.
Variantes genéticas (ClinVar)
266 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 2.116 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Hipoplasia pontocerebelar tipo 2
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Biallelic TSEN2 variants causing pontocerebellar hypoplasia type 2.
Pontocerebellar hypoplasia type 2 (PCH2) is an autosomal recessive neurodegenerative disorder caused by biallelic pathogenic variants in tRNA splicing endonuclease (TSEN) subunit genes. Variants in TSEN54 are most common, with very few cases of TSEN2-related PCH2B reported to date. Here, we report a 7-year-old girl with typical PCH2 features, including progressive microcephaly, epilepsy, developmental delay, cerebellar atrophy, and dystonia. Exome sequencing revealed compound heterozygous TSEN2 variants, a known missense variant NM_025265.4:c.926A>G p.(Tyr309Cys) and a novel nonsense variant c.1048C>T p.(Arg350*). Structural modeling suggested that p.(Tyr309Cys) moderately destabilizes the TSEN2-TSEN54 interface, while p.(Arg350*) truncates the catalytic domain. Despite a minor predicted impact on structure, p.(Tyr309Cys) was associated with severe clinical symptoms in both homozygous and compound heterozygous states. This study expands the TSEN2 mutation spectrum and highlights the utility of integrating structural modeling with clinical data to refine genotype-phenotype correlations in PCH2B.
Spasmodic Abdominal Pain and Other Gastrointestinal Symptoms in Pontocerebellar Hypoplasia Type 2.
Pontocerebellar hypoplasia type 2 (PCH2) is a rare neurodevelopmental disease with a high disease burden. Besides neurological symptoms, somatic symptoms, such as gastroesophageal reflux (GERD) and failure to thrive, are major contributors to this burden. We report three patients with genetically confirmed PCH2A and significant gastrointestinal (GI) symptoms. Apart from impaired swallowing and GERD, which are frequently reported in patients with PCH2, all three patients suffered from episodes of spasmodic abdominal pain and restlessness. In one severely affected patient, lack of intestinal alkaline phosphatase (IAP) is demonstrated. GI symptoms are common in PCH2. We draw attention to episodes of spasmodic abdominal pain seriously, aggravating the condition of the patients, especially their movement disorder, and discuss the role of IAP.
The impact of severe rare chronic neurological disease in childhood on the quality of life of families-a study on MLD and PCH2.
Rare and severe neurological disorders in childhood not only heavily affect the life perspective of the patients, but also their caregivers and families. The aim of this study was to investigate the impact of such diseases on the family, especially on the quality of life and life perspectives of parents, but also on the families' everyday life, based on the model of two diseases which have been well described in recent years with respect to symptoms and course: metachromatic leukodystrophy (MLD) and pontocerebellar hypoplasia type 2 (PCH2). PCH2 is a primary severe developmental disorder, while children with MLD initially develop normally and then progressively deteriorate. Using a semi-standardized questionnaire, 43 families with children suffering from MLD (n = 30) or PCH2 (n = 19) reported data on the severity of the illness/symptoms, on family support and the care situation, as well as on the circumstances of non-affected siblings and the parents' work situation. In addition, the quality of life of parents and general family functioning was assessed using the PedsQL™ Family Impact Module [23]. Results for the latter were compared to published data from families with children without any chronic condition using student's t-tests for independent samples. Potential factors influencing the PedsQL™ scores were analyzed using Spearman's rank correlation. Parents of children with MLD and PCH2 reported significantly lower health-related quality of life (HRQOL) compared to parents of healthy children (P < 0.001). Mothers showed significantly poorer HRQOL (P < 0.05) and were significantly more dissatisfied with their professional development (P < 0.05) than fathers, and this was seen in relation to their child's disease. Neither the form of disease ('primary' symptomatic PCH2 or 'secondary' symptomatic MLD), nor the severity of the child's illness (in terms of gross motor and speech function) had a specific impact on HRQOL in families. However, the time from diagnosis and advanced symptoms in the terminal disease stage were experienced as especially distressing. This study illustrates that MLD and PCH2 affect mothers in particular, but also the entire family. This underlines the need for personalized care and counselling of parents and families, especially following diagnosis and during the end stage in a child with a severe, rare chronic neurological disorder. TSEN54 pontocerebellar hypoplasia (TSEN54-PCH) comprises three PCH phenotypes (PCH2, 4, and 5) that share characteristic neuroradiologic and neurologic findings. The three PCH phenotypes (which differ mainly in life expectancy) were considered to be distinct entities before their molecular basis was known. PCH2. Children usually succumb before age ten years (those with PCH4 and 5 usually succumb as neonates). Children with PCH2 have generalized clonus, uncoordinated sucking and swallowing, impaired cognitive development, lack of voluntary motor development, cortical blindness, and an increased risk for rhabdomyolysis during severe infections. Epilepsy is present in approximately 50%. PCH4. Neonates often have seizures, multiple joint contractures ("arthrogryposis"), generalized clonus, and central respiratory impairment. PCH5 resembles PCH4 and has been described in one family. The diagnosis of TSEN54-PCH is suspected in children with characteristic neuroradiologic and neurologic findings, and is confirmed by the presence of biallelic TSEN54 pathogenic variants. Treatment of manifestations: PCH2: Treatment of irritability, swallowing incoordination, epilepsy, and central visual impairment is symptomatic. Physiotherapy can be helpful. Adequate hydration during prolonged periods of high fever may help avoid rhabdomyolysis. PCH4 and PCH5: No specific therapy is available. Surveillance: PCH2: Routine monitoring of respiratory function, feeding, musculoskeletal and neurologic manifestations, developmental milestones, and family needs. TSEN54-PCH is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a TSEN54 pathogenic variant, each sib of an affected individual has at conception a 25% chance of inheriting both pathogenic variants and being affected, a 50% chance of inheriting one pathogenic variant and being an unaffected carrier, and a 25% chance of inheriting both normal alleles. Once the TSEN54 pathogenic variants have been identified in an affected family member, molecular genetic testing to determine carrier status of at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible.
Levodopa-Responsive Chorea: A Review.
Chorea is one of the disabling movement disorders, and the number of drugs which can treat this disorder effectively is limited. Tetrabenazine and deutetrabenazine are the two drugs approved by the US-FDA for the treatment of chorea associated with HD. Levodopa can improve chorea in some disorders, and this review aims to provide information on the use of levodopa in chorea. A literature search was performed in February 2019 using the following terms "levodopa chorea," "levodopa TITF-1," levodopa brain-lung-thyroid syndrome," and "levodopa Huntington's Disease." The information regarding the etiology, outcome, and dose of levodopa was collected. We found a total of 18 cases in the literature where the benefit was reported with levodopa. Majority of the cases were brain-thyroid-lung (BTL) syndrome (50%). Another 5 cases were HD (Huntington's Disease), one with PCH type 2 (Pontocerebellar hypoplasia type 2), one with meningovascular syphilis, and two patients with Sydenham chorea. The patients with BTL syndrome responded to a very low dose of levodopa. This review suggests that levodopa has the potential to improve chorea in BTL syndrome while its use in chorea due to other disorders requires further study. BTL syndrome due to NKX2-1 mutation responded to levodopa while we did not find any case of chorea due to ADCY-5 mutation responding to levodopa.
TSEN54 Gene-Related Pontocerebellar Hypoplasia Type 2 Could Mimic Dyskinetic Cerebral Palsy with Severe Psychomotor Retardation.
Pontocerebellar hypoplasia (PCH) type 2 is a very rare autosomal recessive neurodegenerative disorder with prenatal onset that disrupts brain development. We present three patients (two siblings and one unrelated child) with PCH 2 linked to the most common mutation c.919G > T (p.Ala307Ser) in TSEN54 gene. The disease started soon after birth with feeding difficulties, extrapyramidal symptoms, psychomotor retardation, progressive microcephaly. Two of the patients were diagnosed with dyskinetic cerebral palsy (CP) at first. Despite the neurodegenerative character of PCH 2, the absence of regression and even some developmental progress in few patients, might erroneously lead to the incorrect diagnosis of dyskinetic CP. Megacisterna magna on brain ultrasound makes the diagnosis of PCH 2 highly probable and should prompt further imaging with MRI. MRI findings of PCH are pivotal for the diagnosis. Genetic testing for the most common mutation in TSEN54 gene should also be performed. Correct diagnosis of PCH 2 is essential not only for the prognosis of the patient, but also for prenatal diagnosis in future pregnancies. Knowledge of the clinical picture of PCH 2 will lead to correct and timely diagnosis. Advanced neuroimaging procedures and molecular genetic techniques provide valuable tools for prompt diagnosis of rare, but clinically important, neurogenetic imitators of CP.
Publicações recentes
Biallelic TSEN2 variants causing pontocerebellar hypoplasia type 2.
Spasmodic Abdominal Pain and Other Gastrointestinal Symptoms in Pontocerebellar Hypoplasia Type 2.
The impact of severe rare chronic neurological disease in childhood on the quality of life of families-a study on MLD and PCH2.
TSEN54 Pontocerebellar Hypoplasia.
Levodopa-Responsive Chorea: A Review.
📚 EuropePMC251 artigos no totalmostrando 10
Biallelic TSEN2 variants causing pontocerebellar hypoplasia type 2.
Journal of human geneticsSpasmodic Abdominal Pain and Other Gastrointestinal Symptoms in Pontocerebellar Hypoplasia Type 2.
NeuropediatricsThe impact of severe rare chronic neurological disease in childhood on the quality of life of families-a study on MLD and PCH2.
Orphanet journal of rare diseasesLevodopa-Responsive Chorea: A Review.
Annals of Indian Academy of NeurologyTSEN54 Gene-Related Pontocerebellar Hypoplasia Type 2 Could Mimic Dyskinetic Cerebral Palsy with Severe Psychomotor Retardation.
Frontiers in pediatricsBrain morphometry in Pontocerebellar Hypoplasia type 2.
Orphanet journal of rare diseasesMilder progressive cerebellar atrophy caused by biallelic SEPSECS mutations.
Journal of human geneticsTSEN54 gene-related pontocerebellar hypoplasia type 2 presenting with exaggerated startle response: report of two cases in a family.
The Turkish journal of pediatricsA combination of chorea, myoclonus, and dystonia in a patient with pontocerebellar hypoplasia type 2: a video case presentation.
Acta neurologica BelgicaTargeted carrier screening for four recessive disorders: high detection rate within a founder population.
European journal of medical geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Hipoplasia pontocerebelar tipo 2.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Hipoplasia pontocerebelar tipo 2
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Biallelic TSEN2 variants causing pontocerebellar hypoplasia type 2.
- Spasmodic Abdominal Pain and Other Gastrointestinal Symptoms in Pontocerebellar Hypoplasia Type 2.
- The impact of severe rare chronic neurological disease in childhood on the quality of life of families-a study on MLD and PCH2.
- Levodopa-Responsive Chorea: A Review.
- TSEN54 Gene-Related Pontocerebellar Hypoplasia Type 2 Could Mimic Dyskinetic Cerebral Palsy with Severe Psychomotor Retardation.
- TSEN54 Pontocerebellar Hypoplasia.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2524(Orphanet)
- MONDO:0016759(MONDO)
- GARD:10705(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q2195280(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
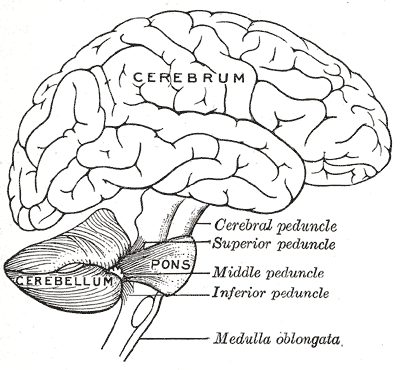
Hipoplasia pontocerebelar tipo 2
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata