Obstrução congênita do ducto nasolacrimal é uma condição rara de herança autossômica recessiva, caracterizada pela atresia do ducto lacrimal. Manifesta-se com epífora, conjuntivite, edema periorbital, e pode levar a dacriocistocele, sinusite ou dacriocistite.
Introdução
O que você precisa saber de cara
Visão geral
A obstrução congênita do ducto nasolacrimal é uma condição rara que afeta o sistema de drenagem das lágrimas, presente desde o nascimento ou antes do nascimento. Estima-se que sua prevalência seja inferior a 1 caso por 1.000.000 de pessoas. A condição é herdada de forma autossômica recessiva, ou seja, é necessário que ambos os pais transmitam uma cópia do gene alterado para que a criança manifeste a doença.[1][4]
Sinais e sintomas
Os principais sinais e sintomas incluem epífora (lacrimejamento excessivo), dacriocistite (inflamação do saco lacrimal), dacriocistocele (cisto no ducto lacrimal), conjuntivite, sinusite, edema periorbital (inchaço ao redor dos olhos) e atresia do ducto lacrimal (ausência ou fechamento anormal do ducto). Esses sintomas podem se manifestar já no período neonatal ou mesmo antes do nascimento.[1][4]
Causas genéticas
Diagnóstico
O diagnóstico pode ser confirmado por meio de testes genéticos. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para procedimentos como cariótipo com bandas G, Q ou R, pesquisa de microdeleções/microduplicações por FISH, sequenciamento completo do exoma (WES) e dosagem de alfa-fetoproteína. Atualmente, há 15 variantes registradas no ClinVar associadas a esta condição, e 3 tipos de testes genéticos disponíveis.[1][2][5]
Tratamento e manejo
O manejo da obstrução congênita do ducto nasolacrimal é individualizado e deve ser conduzido por uma equipe multidisciplinar. O SUS oferece atendimento em reabilitação para doenças raras como parte do suporte. Não há medicamentos específicos aprovados para esta condição; o tratamento é geralmente cirúrgico ou de suporte, dependendo da gravidade dos sintomas. Consulte um médico especialista para orientação personalizada.[1][2]
Prognóstico e qualidade de vida
O prognóstico depende da gravidade da obstrução e da presença de complicações como infecções recorrentes (dacriocistite, sinusite). Com o manejo adequado, incluindo intervenções cirúrgicas quando necessário, a maioria das crianças pode ter boa qualidade de vida. O acompanhamento regular com oftalmologista e geneticista é recomendado.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Obstrução congênita do ducto nasolacrimal é uma condição rara de herança autossômica recessiva, caracterizada pela atresia do ducto lacrimal. Manifesta-se com epífora, conjuntivite, edema periorbital, e pode levar a dacriocistocele, sinusite ou dacriocistite.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A obstrução congênita do ducto nasolacrimal é uma condição rara que afeta o sistema de drenagem das lágrimas, presente desde o nascimento ou antes do nascimento. Estima-se que sua prevalência seja inferior a 1 caso por 1.000.000 de pessoas. A condição é herdada de forma autossômica recessiva, ou seja, é necessário que ambos os pais transmitam uma cópia do gene alterado para que a criança manifeste a doença.[1][4]
Sinais e sintomas
Os principais sinais e sintomas incluem epífora (lacrimejamento excessivo), dacriocistite (inflamação do saco lacrimal), dacriocistocele (cisto no ducto lacrimal), conjuntivite, sinusite, edema periorbital (inchaço ao redor dos olhos) e atresia do ducto lacrimal (ausência ou fechamento anormal do ducto). Esses sintomas podem se manifestar já no período neonatal ou mesmo antes do nascimento.[1][4]
Causas genéticas
Diagnóstico
O diagnóstico pode ser confirmado por meio de testes genéticos. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para procedimentos como cariótipo com bandas G, Q ou R, pesquisa de microdeleções/microduplicações por FISH, sequenciamento completo do exoma (WES) e dosagem de alfa-fetoproteína. Atualmente, há 15 variantes registradas no ClinVar associadas a esta condição, e 3 tipos de testes genéticos disponíveis.[1][2][5]
Tratamento e manejo
O manejo da obstrução congênita do ducto nasolacrimal é individualizado e deve ser conduzido por uma equipe multidisciplinar. O SUS oferece atendimento em reabilitação para doenças raras como parte do suporte. Não há medicamentos específicos aprovados para esta condição; o tratamento é geralmente cirúrgico ou de suporte, dependendo da gravidade dos sintomas. Consulte um médico especialista para orientação personalizada.[1][2]
Prognóstico e qualidade de vida
O prognóstico depende da gravidade da obstrução e da presença de complicações como infecções recorrentes (dacriocistite, sinusite). Com o manejo adequado, incluindo intervenções cirúrgicas quando necessário, a maioria das crianças pode ter boa qualidade de vida. O acompanhamento regular com oftalmologista e geneticista é recomendado.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 6 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 8 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Membrane
Lacrimal duct defect
A condition resulting in the imbalance between tear production and tear drainage. Infants typically manifest persistent epiphora and/or recurrent infections of the lacrimal pathway, such as conjunctivitis. LCDD is caused by failure of the nasolacrimal duct to open into the inferior meatus.
Variantes genéticas (ClinVar)
15 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 3 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Obstrução congênita do ducto nasolacrimal
Centros de Referência SUS
24 centros habilitados pelo SUS para Obstrução congênita do ducto nasolacrimal
Centros para Obstrução congênita do ducto nasolacrimal
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Congenital nasolacrimal duct obstruction outcomes.
To characterize children with congenital nasolacrimal duct obstruction (NLDO) seen by a pediatric ophthalmologist in terms of initiation of tear duct massage and need for lacrimal duct probing. Retrospective chart review. All children with a diagnosis of congenital NLDO managed by one pediatric ophthalmologist (SH) at a tertiary pediatric center (McMaster Children's Hospital, Hamilton, Ontario, Canada). Charts were reviewed to determine the percentage of patients requiring nasolacrimal duct probing. Secondary outcomes were percentage of patients who had tear duct massage, accurate or inaccurate, initiated by the referring provider. Overall, 113 patients with a mean age of 1.8 ± 1.5 years at presentation were included, and 55 (48.7%) were male. Most patients (83.2%) were referred by a primary care provider (family physician or pediatrician) and the remainder by optometrists (13.3%) or ophthalmologists (3.5%). Accurate tear duct massage was initiated by referring providers in 23% of cases. Ultimately, 44.2% of cases required probing, and 4.4% were still pending follow-up at time of data collection. Of children who learned accurate massage technique, 56% avoided surgical intervention, even when proper massage is introduced after 12 months of age. Most patients referred to pediatric ophthalmology for congenital NLDO did not have an appropriate trial of tear duct massage before referral, and less than half of patients required probing after appropriate massage was initiated. Educating primary care providers and optometrists on proper initiation of tear duct massage may reduce the volume of congenital NLDO referrals to pediatric ophthalmology, a subspecialty with limited physicians and long wait times.
FGF10/IGSF3 Variants in Bony Congenital Nasolacrimal Duct Obstruction: A Genotype-Phenotype Study of Syndromic Versus Isolated Disease.
The purpose of this study was to identify pathogenic variants associated with congenital bony nasolacrimal duct obstruction (bony CNLDO) and to clarify genotype-phenotype correlations. In this single-center retrospective case study, children with clinically confirmed bony CNLDO and their relatives underwent detailed ophthalmic, systemic examinations, orbital computed tomography (CT), and maxillofacial magnetic resonance imaging (MRI). Whole-exome sequencing (WES) was performed on peripheral blood samples. Bioinformatics tools were utilized to predict variant pathogenicity, and classifications were performed according to the American College of Medical Genetics and Genomics (ACMG) guidelines. Thirty-two participants were included in this study, comprising six families and seven sporadic cases. WES identified 8 novel FGF10 variants, including 6 missense mutations (c.598C>G, c.316T>G, c.395T>C, c.316T>C, c.327C>G, and c.332T>G), one intronic splicing mutation (c.429+2T>A), and one heterozygous deletion at chromosome 5p12 encompassing the FGF10 gene. Two sporadic cases had no identifiable pathogenic variants. All patients with FGF10 variants exhibited syndromic aplasia of lacrimal and salivary glands (ALSGs) phenotype characterized by bony CNLDO, punctal anomalies, and hypoplasia of lacrimal, parotid, and submandibular glands. Three novel IGSF3 variants were also identified including two missense mutations (c.2872G>C and c.2531G>A) and one nonsense mutation (c.2416G>T). In contrast, individuals carrying IGSF3 variants showed isolated bony CNLDO with normal glandular morphology. Bony CNLDO is predominantly a monogenic disorder involving FGF10 or IGSF3. FGF10 mutations are associated with syndromic, multi-gland hypoplasia, whereas IGSF3 mutations result in isolated bony CNLDO. These findings enhance understanding of the genetic heterogeneity and genotype-phenotype correlations underlying bony CNLDO, with real-world implications for genetic diagnosis and counseling.
Outcomes of Eye Examination and Vision Screening in Term Infants Presenting to a Tertiary Hospital in Türkiye.
Ophthalmic screening is an important part of the medical care of children as some eye abnormalities can lead to irreversible vision loss if not treated in the first few months or years of life. The aim of this study is to evaluate the outcomes of the ophthalmic screening program in term infants aged ≤1 year who presented to a tertiary hospital in Türkiye. The records of 1,035 infants ≤1 year old who underwent ophthalmic screening between November 2019 and February 2022 were reviewed retrospectively. Demographic and medical details, parental complaints about the infants' eyes, family history of ocular, adnexal, and systemic pathologies, light reactions, red reflex test results, eye movements, blink response to light, fixation and following, noticeable strabismus, conjunctivitis, epiphora, anterior segment and fundus pathologies, and treatments applied were recorded. The referring physician (family physician, pediatrician) and reason for reference were also noted. Abnormal ophthalmological findings were detected in 136 infants (13.14%). The most common finding was congenital nasolacrimal duct obstruction (72.05%), followed by strabismus (8.82%), ptosis (4,41%), absence of following (3.67%), congenital cataract (2.94%), hemangioma of the adnexa (2.94%), nystagmus (2.94%), albino fundus (1.47%), preretinal hemorrhage (1.47%), and coloboma of the iris and choroid (1.47%). We detected abnormal red reflex in 4 infants who were not referred for red reflex abnormality by the referring physician, while another 4 infants referred for red reflex abnormality had no pathology on ocular examinations including the red reflex test. The importance of ophthalmic screening in infants is well appreciated but there are inadequacies in performing and interpreting the red reflex test among family physicians and pediatricians. Efforts should be directed at improving vision screening skills, especially red reflex testing. Çeşitli göz hastalıkları, yaşamın ilk birkaç ayı ya da yılında tedavi edilmezlerse geri dönüşü olmayan görme kaybına yol açabilmektedir. Bu nedenle göz taraması çocukların genel tıbbi muayenesinin önemli bir parçasıdır. Bu çalışmanın amacı Türkiye’de üçüncü basamak hastaneye başvuran 1 yaş ve altı term bebeklerde başvuru nedenlerini ve göz tarama programının sonuçlarını değerlendirmektir. Kasım 2019-Şubat 2022 tarihleri arasında göz taraması yapılan 1 yaş ve altı 1.035 bebeğin kayıtları geriye dönük olarak incelendi. Demografik ve medikal veriler, varsa anne-babanın bebeğinin gözüyle ilgili şikayetleri, oküler, sistemik ve adneksiyal patolojilerle ilgili aile öyküsü, ışık reaksiyonları, kırmızı refle testi, göz hareketleri, ışığa kırpma refleksi, fiksasyon, takip, strabismus, konjonktivit, sulanma, ön segment ve fundus patolojileri ile uygulanan tedaviler not edildi. Hastayı sevk eden hekim (aile hekimi, çocuk doktoru) ve sevk nedeni kaydedildi. Bebeklerin 136’sında (%13,14) anormal oftalmolojik bulgular saptandı. En sık konjenital nazolakrimal kanal tıkanıklığı (%72,05) görülürken, bunu şaşılık (%8,82), ptozis (%4,41), göz takibi olmaması (%3,67), konjenital katarakt (%2,94), adneksiyal hemanjiom (%2,94), nistagmus (%2,94), albinoid fundus (%1,47), preretinal hemoraji (%1,47) ve iris ve koroid kolobomu (%1,47) takip etmekteydi. Muayene sırasında 4 bebekte anormal kırmızı refle testi mevcut iken bu beklerin hiçbirinin sevk nedeni anormal kırmızı refle testi değildi. Ayrıca kırmızı refle patolojisi ile sevk edilen 4 bebeğin ise kırmızı refle testi dahil olmak üzere oftalmolojik muayenelerinde patoloji yoktu. Bebeklerde göz taramasının önemi çok iyi bilinmekle beraber aile hekimleri ve pediatristler arasında kırmızı refle testinin yorumlanmasında farklılıklar olabilmektedir. Bu konudaki çalışmalar özellikle kırmızı refle testinin uygulanmasının ve yorumlanmasının geliştirilmesi üzerinde yoğunlaştırılmalıdır.
Relation between mode of delivery and related factors with congenital nasolacrimal duct obstruction.
The purpose of the study was to evaluate the possible relation between the mode of delivery and associated factors with congenital nasolacrimal duct obstruction (CNLDO). This case-control study was conducted on children between 6 months and 5 years with CNLDO and healthy controls. A binary logistic regression model was fitted to identify the associated factors with CNLDO and a backward elimination technique was applied for modeling. From 324 enrolled subjects, 160 were as a case (CNLDO group) and 164 were healthy control (non-CNLDO group). A positive family history of CNLDO was present in 21 (13.1%) patients with CNLDO and 3 (1.81%) controls (P < 0.001). Cesarean section was 62.9% and 64.0% in the CNLDO and controls groups, respectively (P = 0.83). Using logistic regression models, children with a positive family history were 10.12 times more likely to have CNLDO than the control group (odds ratio [OR] = 10.12, 95% confidence interval [CI] 2.838-36.069). In addition, birth weight ≤2500 g (OR = 2.39, 95% CI: (1.123-5.087)) and maternal age ≤27 years at the time of delivery (OR = 2.35, 95% CI: (1.462-3.778)) were associated with upper odds for CNLDO. There is an increase in the risk of CNLDO with a positive family history, birth weight < 2500 g, and maternal age of <27 years. However, further research is warranted to evaluate the causal relationship of these risk factors. Notably, there is no significant relationship between the mode of delivery and developing CNLDO.
A Family with EEC Syndrome in the Son and ADULT Syndrome in His Father Caused by the c.797G>A (p.Arg266Gln) Pathogenic Variant in the TP63 Gene.
To our knowledge, there are few examples of intrafamilial variability involving two different TP63-linked morphopathies within a same family. Here, we describe a Mexican family in which the son had ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome 3 (EEC3), and his father acro-dermato-ungual-lacrimal-tooth (ADULT) syndrome, both heterozygous for the p.Arg266Gln pathogenic variant in TP63. Additionally, we reviewed the clinical information reported for this TP63 genotype. The son of this family presented ectodermal defects (thin and sparse hair, mild nail dysplasia), tetramelic ectrodactyly, syndactyly, and nasolacrimal duct obstruction (NLDO), indicative of an EEC3 diagnosis. His father, however, exhibited severe NLDO, facial freckling, dental abnormalities, mild nail dysplasia, and a history of micturition problems, compatible with ADULT syndrome. Both were heterozygous for the NM_003722.5(TP63):c.797G>A (p.Arg266Gln) pathogenic variant in TP63. This report expands the spectrum of intrafamilial variability confirming that this can include the expression of distinct types of TP63-related disorders among different members of the same family, whose implications should be also considered in genetic counseling. From our review, we observed that p.Arg266Gln variant seems to correlate particularly with the presence of NLDO, sparse hair/eyebrows, ridged/dystrophic nails, anodontia/hypodontia, and micturition difficulties, as well as for a minor frequency of cleft lip/cleft palate.
Publicações recentes
FGF10/IGSF3 Variants in Bony Congenital Nasolacrimal Duct Obstruction: A Genotype-Phenotype Study of Syndromic Versus Isolated Disease.
A spectrum of TP63-related disorders with eight affected individuals in five unrelated families.
Bony Congenital Nasolacrimal Duct Obstruction: A Novel Phenotype of Aplasia of Lacrimal and Major Salivary Glands.
Absent meibomian glands and cone dystrophy in ADULT syndrome: identification by whole exome sequencing of pathogenic variants in two causal genes TP63 and CNGB3.
The Evolving Story of CNLDO: Serial Photographic Documentation and Parental Perspectives.
📚 EuropePMC1 artigos no totalmostrando 26
Congenital nasolacrimal duct obstruction outcomes.
Paediatrics & child healthFGF10/IGSF3 Variants in Bony Congenital Nasolacrimal Duct Obstruction: A Genotype-Phenotype Study of Syndromic Versus Isolated Disease.
Investigative ophthalmology & visual scienceOutcomes of Eye Examination and Vision Screening in Term Infants Presenting to a Tertiary Hospital in Türkiye.
Turkish journal of ophthalmologyRelation between mode of delivery and related factors with congenital nasolacrimal duct obstruction.
Saudi journal of ophthalmology : official journal of the Saudi Ophthalmological SocietyA Family with EEC Syndrome in the Son and ADULT Syndrome in His Father Caused by the c.797G>A (p.Arg266Gln) Pathogenic Variant in the TP63 Gene.
Molecular syndromologyA spectrum of TP63-related disorders with eight affected individuals in five unrelated families.
European journal of medical geneticsFamilial Occurrence of Isolated Late-onset Nasolacrimal Duct Obstruction in Two Unrelated Families.
Rambam Maimonides medical journalBony Congenital Nasolacrimal Duct Obstruction: A Novel Phenotype of Aplasia of Lacrimal and Major Salivary Glands.
OphthalmologyAbsent meibomian glands and cone dystrophy in ADULT syndrome: identification by whole exome sequencing of pathogenic variants in two causal genes TP63 and CNGB3.
Ophthalmic geneticsIdentification of a novel mutation in the FGF10 gene in a Chinese family with obvious congenital lacrimal duct dysplasia in lacrimo-auriculo-dento-digital syndrome.
International journal of ophthalmologyThe Evolving Story of CNLDO: Serial Photographic Documentation and Parental Perspectives.
Ophthalmic plastic and reconstructive surgeryA case of blepharophimosis: Freeman Sheldon syndrome.
Ophthalmic geneticsEfficacy and Safety of Inhalation Sedation during Office Probing for Congenital Nasolacrimal Duct Obstruction.
Journal of clinical medicineOffice- or Facility-Based Probing for Congenital Nasolacrimal Duct Obstruction: A Report by the American Academy of Ophthalmology.
Ophthalmology[Congenital nasolacrimal duct obstruction : A real-life study from the first symptoms to the results of surgical treatment].
Der Ophthalmologe : Zeitschrift der Deutschen Ophthalmologischen GesellschaftPreliminary report on screening IGSF3 gene mutation in families with congenital absence of lacrimal puncta and canaliculi.
International journal of ophthalmologyLate complications of congenital nasolacrimal duct obstruction in a Taiwanese family.
Pediatrics and neonatologyIs there an association between congenital nasolacrimal duct obstruction and cesarean delivery?
European journal of ophthalmologyPrevalence of childhood ocular morbidity in a peri-urban setting in Bangladesh: a community-based study.
Public healthADULT syndrome: phenotype in a Brazilian family with the R298Q mutation.
International journal of dermatologyUniversal ocular screening of 481 infants using wide-field digital imaging system.
BMC ophthalmologyPrevalence and Risk Factors of Amblyopia among Refractive Errors in an Eastern European Population.
Medicina (Kaunas, Lithuania)Familial Incomplete Punctal Canalization: Clinical and Fourier Domain Optical Coherence Tomography Features.
Ophthalmic plastic and reconstructive surgeryAcro-Dermato-Ungual-Lacrimal-Tooth Syndrome: An Uncommon Member of the Ectodermal Dysplasias.
Pediatric dermatology[Treatment of congenital lacrimal duct obstruction: A prospective clinical cohort study].
Der Ophthalmologe : Zeitschrift der Deutschen Ophthalmologischen GesellschaftUnusual manifestations of ectodermal dysplasia-syndactyly syndrome type I in two Yemeni siblings.
Dermatology online journalAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Obstrução congênita do ducto nasolacrimal.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Obstrução congênita do ducto nasolacrimal
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Congenital nasolacrimal duct obstruction outcomes.
- FGF10/IGSF3 Variants in Bony Congenital Nasolacrimal Duct Obstruction: A Genotype-Phenotype Study of Syndromic Versus Isolated Disease.
- Outcomes of Eye Examination and Vision Screening in Term Infants Presenting to a Tertiary Hospital in Türkiye.
- Relation between mode of delivery and related factors with congenital nasolacrimal duct obstruction.Saudi journal of ophthalmology : official journal of the Saudi Ophthalmological Society· 2025· PMID 41179374mais citado
- A Family with EEC Syndrome in the Son and ADULT Syndrome in His Father Caused by the c.797G>A (p.Arg266Gln) Pathogenic Variant in the TP63 Gene.
- A spectrum of TP63-related disorders with eight affected individuals in five unrelated families.
- Bony Congenital Nasolacrimal Duct Obstruction: A Novel Phenotype of Aplasia of Lacrimal and Major Salivary Glands.
- Absent meibomian glands and cone dystrophy in ADULT syndrome: identification by whole exome sequencing of pathogenic variants in two causal genes TP63 and CNGB3.
- The Evolving Story of CNLDO: Serial Photographic Documentation and Parental Perspectives.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:451612(Orphanet)
- OMIM OMIM:149700(OMIM)
- MONDO:0007871(MONDO)
- GARD:17784(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55950244(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
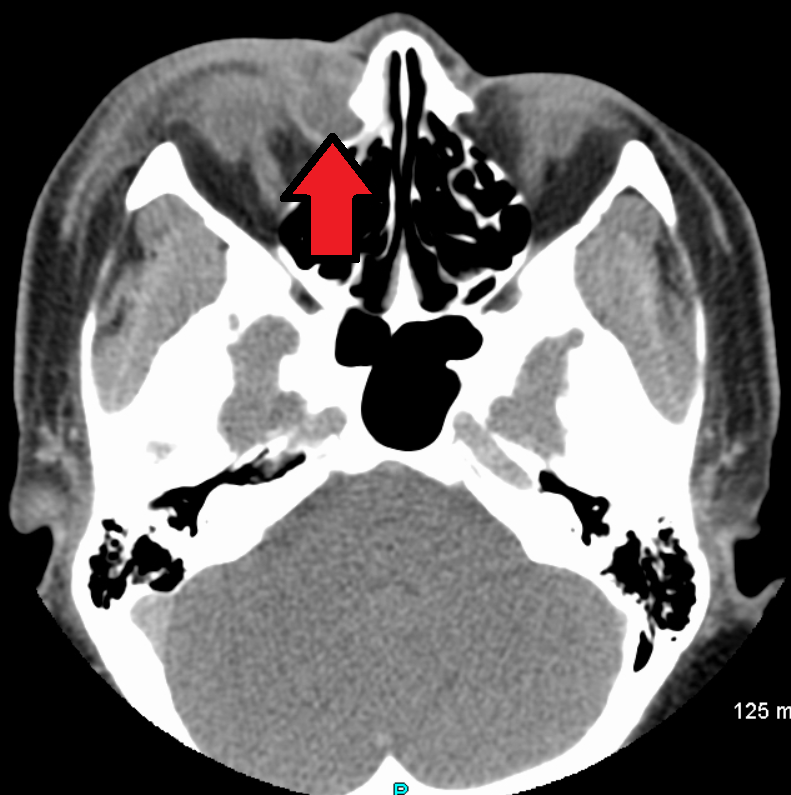
Obstrução congênita do ducto nasolacrimal
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata