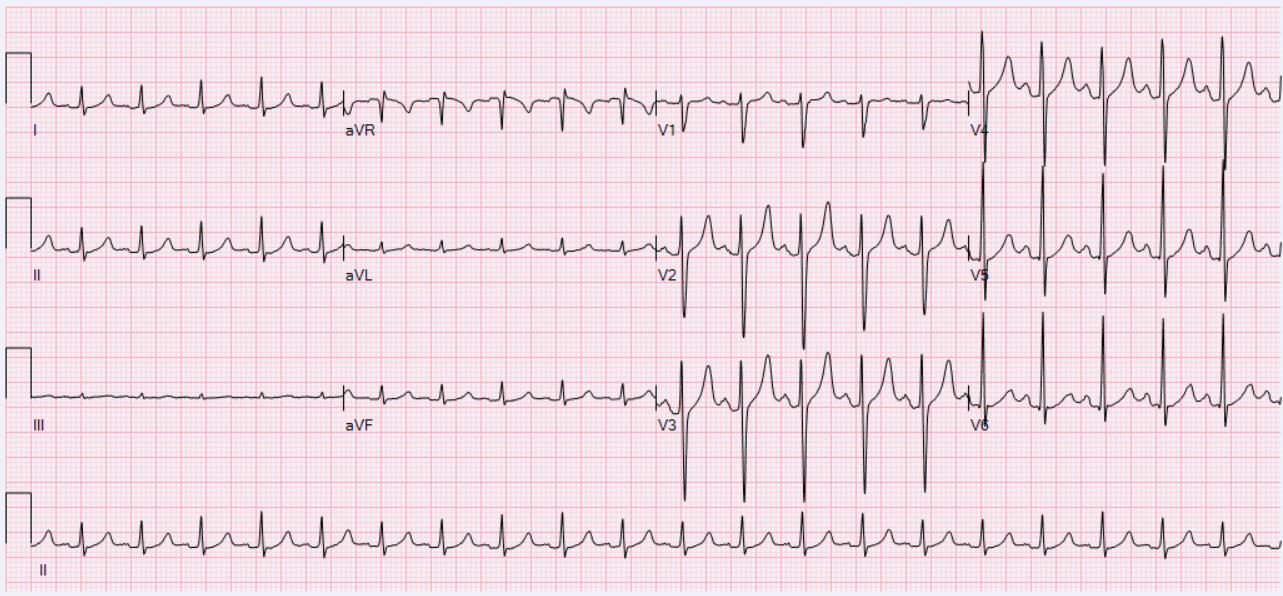
A síndrome do nó sinusal é uma doença rara do ritmo cardíaco, geralmente em idosos, caracterizada por achados eletrocardiográficos de bradicardia sinusal, fibrilação atrial, taquicardia atrial, parada sinusal ou bloqueio sinoatrial, e que se manifesta com sintomas como síncope, tontura, palpitações, fadiga ou mesmo insuficiência cardíaca. Resulta do mau funcionamento do sistema de condução cardíaca, provavelmente secundário à fibrose degenerativa do tecido nodal em idosos ou secundário a distúrbios cardíacos em pacientes mais jovens.
Introdução
O que você precisa saber de cara
A síndrome do nó sinusal é uma doença rara do ritmo cardíaco, geralmente em idosos, caracterizada por achados eletrocardiográficos de bradicardia sinusal, fibrilação atrial, taquicardia atrial, parada sinusal ou bloqueio sinoatrial, e que se manifesta com sintomas como síncope, tontura, palpitações, fadiga ou mesmo insuficiência cardíaca. Resulta do mau funcionamento do sistema de condução cardíaca, provavelmente secundário à fibrose degenerativa do tecido nodal em idosos ou secundário a distúrbios cardíacos em pacientes mais jovens.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 11 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 29 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
4 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive.
Muscle contraction
Cytoplasm, myofibril
Atrial septal defect 3
A congenital heart malformation characterized by incomplete closure of the wall between the atria resulting in blood flow from the left to the right atria.
Hyperpolarization-activated ion channel that are permeable to Na(+) and K(+) ions with very slow activation and inactivation (PubMed:10228147, PubMed:10430953, PubMed:20829353). Exhibits higher selectivity for K(+) over Na(+) ions (PubMed:10228147). Contributes to the native pacemaker currents in heart (If) that regulate the rhythm of heart beat (Probable) (PubMed:10228147, PubMed:16407510, PubMed:19165230). Contributes to the native pacemaker currents in neurons (Ih) (Probable). May mediate res
Cell membrane
Sick sinus syndrome 2
The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth.
Pore-forming subunit of Nav1.5, a voltage-gated sodium (Nav) channel that directly mediates the depolarizing phase of action potentials in excitable membranes. Navs, also called VGSCs (voltage-gated sodium channels) or VDSCs (voltage-dependent sodium channels), operate by switching between closed and open conformations depending on the voltage difference across the membrane. In the open conformation they allow Na(+) ions to selectively pass through the pore, along their electrochemical gradient.
Cell membraneCytoplasm, perinuclear regionCell membrane, sarcolemma, T-tubuleCell junction
Progressive familial heart block 1A
A cardiac bundle branch disorder characterized by progressive alteration of cardiac conduction through the His-Purkinje system, with a pattern of a right bundle-branch block and/or left anterior hemiblock occurring individually or together. It leads to complete atrio-ventricular block causing syncope and sudden death.
Guanine nucleotide-binding proteins (G proteins) are involved as a modulator or transducer in various transmembrane signaling systems. The beta and gamma chains are required for the GTPase activity, for replacement of GDP by GTP, and for G protein-effector interaction
Cytoplasm, perinuclear regionCell membrane
Neurodevelopmental disorder with hypotonia and dysmorphic facies
An autosomal dominant disorder characterized by global developmental delay, hypotonia, and variably impaired intellectual development, often with speech delay and delayed walking. Most patients have dysmorphic facial features. Clinical features are highly variable and may include congenital cardiac defects, non-specific renal anomalies, joint contractures or joint hyperextensibility, dry skin, and cryptorchidism.
Variantes genéticas (ClinVar)
1.846 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
33 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome do nó sinusal doente hereditária
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Mostrando amostra de 16 publicações de um total de 832
Systematic analysis of SCN5A variants associated with inherited cardiac diseases.
SCN5A variants are associated with a spectrum of cardiac electrical disorders with clear phenotypes. However, they may also be associated with complex phenotypic traits like overlap syndromes or pleiotropy, which have not been systematically described. In addition, the involvement of SCN5A in dilated cardiomyopathies (DCMs) remains controversial. We aimed to evaluate the different phenotypes associated with pathogenic (P)/likely pathogenic (LP) SCN5A variants and to determine the prevalence of pleiotropy in a large multicentric cohort of P/LP SCN5A variant carriers. The DNA of 13,510 consecutive probands (9960 with cardiomyopathies) was sequenced with a custom panel of genes. Individuals carrying a heterozygous single P/LP SCN5A variant were selected and phenotyped. The study included 170 P/LP variants found in 495 patients. Of them, 119 (70%) were exclusively associated with a single well-established phenotype: 91 with Brugada syndrome, 15 with type 3 long QT syndrome, 6 with progressive cardiac conduction disease, 4 with multifocal ectopic Purkinje-related premature contractions, and 3 with sick sinus syndrome. Thirty-two variants (19%) were associated with overlap syndromes or pleiotropy. The 19 remaining variants (11%) were associated with atypical or unclear phenotypes. Of those, 8 were carried by 8 patients presenting with DCM with a debatable causative genotype/phenotype link. Most P/LP SCN5A variants were found in patients with primary electrical disorders, mainly Brugada syndrome. Nearly 20% were associated with overlap syndromes or pleiotropy, underscoring the need for comprehensive phenotypic evaluation. The concept of SCN5A variants causing DCM is extremely rare (8/9960) if not questionable. Hereditary transthyretin amyloidosis (ATTRv amyloidosis) is characterized by a slowly progressive peripheral sensorimotor and/or autonomic neuropathy. Amyloidosis can involve the heart, central nervous system (CNS), eyes, and kidneys. The disease usually begins in the third to fifth decade in persons from endemic foci in Portugal and Japan; onset is later in persons from other areas. Typically, sensory neuropathy starts in the lower extremities with paresthesia and hypesthesia of the feet, followed within a few years by motor neuropathy. In some persons, particularly those with early-onset disease, autonomic neuropathy is the first manifestation of the condition; findings can include orthostatic hypotension, constipation alternating with diarrhea, attacks of nausea and vomiting, delayed gastric emptying, sexual impotence, anhidrosis, and urinary retention or incontinence. Cardiac amyloidosis is mainly characterized by progressive restrictive cardiomyopathy. Individuals with leptomeningeal amyloidosis may have the following CNS findings: dementia, psychosis, visual impairment, headache, seizures, motor paresis, ataxia, myelopathy, hydrocephalus, or intracranial hemorrhage. Ocular involvement includes vitreous opacity, glaucoma, dry eye, and ocular amyloid angiopathy. Mild-to-severe kidney disease can develop. The diagnosis of ATTRv amyloidosis is established in a proband with characteristic clinical features, including imaging or histopathology findings of amyloidosis, and a heterozygous pathogenic variant in TTR identified by molecular genetic testing. Targeted therapies: Pharmacotherapeutics (e.g., gene-silencing therapies, transthyretin tetramer stabilizers) are first-line therapy for all individuals with ATTRv amyloidosis. There is limited indication for orthotopic liver transplantation. Treatment of manifestations: Pharmacologic treatments for neuropathic pain; surgical release for carpal tunnel syndrome; ankle-foot orthoses and physical therapy for motor neuropathy; standard treatments for autonomic dysfunction and CNS manifestations. In those with sick sinus syndrome or second- or third-degree atrioventricular block, a cardiac pacemaker may be indicated. Vitrectomy for vitreous opacification; surgical treatment for glaucoma; ocular lubrication for dry eye; erythropoietin or intravenous iron for normocytic normochromic anemia; hemodialysis as needed for end-stage kidney disease. Surveillance: Abdominal wall fat aspiration or gastrointestinal tract biopsy annually to identify disease onset in asymptomatic individuals; systematic neurologic screening at least annually; nerve conduction studies annually; clinical assessment for manifestations of cardiac disease and serum B-type natriuretic peptide levels annually; electrocardiogram and echocardiography at least annually; 99mTc-PYP myocardial scintigraphy every three to five years; clinical assessment for dementia, psychosis, headache, seizures, motor paresis, and ataxia annually; ophthalmology examination including assessment for glaucoma at least annually; laboratory assessment of kidney function annually; modified body mass index annually; assessment of psychological manifestations as needed. Agents/circumstances to avoid: Local heating appliances, such as hot-water bottles, which can cause low-temperature burn injuries in those with decreased temperature and pain perception. Evaluation of relatives at risk: Clarify the genetic status of at-risk relatives by molecular genetic testing for the TTR pathogenic variant(s) in the family in order to identify as early as possible those who would benefit from prompt early diagnosis and treatment. ATTRv amyloidosis is inherited in an autosomal dominant manner. Each child of an individual who is heterozygous for a TTR pathogenic variant has a 50% risk of inheriting the TTR pathogenic variant. All offspring of an individual who has biallelic TTR pathogenic variants will inherit a pathogenic variant. Once the TTR pathogenic variant(s) has been identified in an affected family member, predictive testing for at-risk family members and prenatal/preimplantation genetic testing are possible.
Adult late-onset limb-girdle muscular dystrophy R1/2A complicated by parathyroid adenoma and sick sinus syndrome: a case report and literature review.
Limb-girdle muscular dystrophy (LGMD) is a group of hereditary myopathies. This group of diseases is highly heterogeneous in terms of genetic mode, age at onset, and disease progression; therefore, they are easily misdiagnosed and missed in clinical practice. We describe a case of adult late-onset LGMD R1/2A in a 56-year-old female patient. The patient experienced elevated creatine kinase levels lasting 5 years, muscle soreness of the limbs lasting 4 years, and exacerbation of limb fatigue lasting 1 month. Early in the course of the disease, the patient experienced severe bradycardia and was later diagnosed with sick sinus syndrome. In addition to cardiac involvement, our patient also had primary hyperparathyroidism during the disease course, which was confirmed pathologically as a parathyroid adenoma. A biopsy of the left biceps showed pathological manifestations of mild myogenic damage. All-exon gene sequencing confirmed the diagnosis of LGMD R1/2A, and she was treated with vitamin E, vitamin B2, and coenzyme Q. Due to atrial fibrillation secondary to sick sinus syndrome, a pacemaker was implanted. The patient in this case study had adult late-onset LGMD R1/2A with cardiac involvement and functional parathyroid adenoma, which is rare and clinically significant. Therefore, early clinical identification, diagnosis, as well as targeted and active treatments can improve the prognosis of such patients.
Incidence and predictors of pacemaker implantation at follow-up after reversible high-degree sinus node dysfunction or atrioventricular block.
A pacemaker implantation is not indicated in cases of reversible high-degree symptomatic sinus node dysfunction (SND) and atrioventricular block (AVB). However, it remains uncertain whether these reversible automaticity/conduction disorders may recur in some patients at follow-up, in the absence of reversible cause. This retrospective study aimed to determine the incidence and predictive factors of permanent pacemaker (PPM) implantation at follow-up and after reversible high-degree SND/AVB. Based on medical electronic files codes, we identified patients who were hospitalized in our cardiac intensive care unit between January 2003 and December 2020 due to reversible high-degree SND/AVB and who were discharged from the hospital alive and without PPM implantation. Acute myocardial infarction and post-cardiac surgery patients were excluded. We categorized the patients according to the need for PPM at follow-up due to non-reversible high-degree SND/AVB. Of the 93 patients included, 26 patients (28%) were readmitted for PPM implantation at follow-up after hospital discharge. Among baseline characteristics, compared with patients who did not have high-degree SND/AVB recurrence, those who had subsequent PPM implantation had less frequent previous hypertension (70% vs. 46%, p = .031). Regarding the initial causes of reversible SND/AVB, isolated hyperkalemia was found more often in the patients readmitted for PPM (19% vs. 3% vs. p = .017). Moreover, recurrence of high-degree SND/AVB was significantly associated with the presence of intraventricular conduction disorders (either bundle branch block or left bundle branch hemiblock) on ECG at discharge (36% in patients without PPM vs. 68% in PPM patients, p = .012). Almost one third of the patients discharged alive from the hospital after a reversible high-degree SND/AVB needed a pacemaker implantation at follow-up. Complete bundle branch block or left bundle branch hemiblock on discharge ECG after recovery of atrioventricular conduction and/or sinus automaticity was associated with a greater risk of recurrence leading to pacemaker implantation.
[Clinical case of the cardiovascular system involvement in a patient with Charcot-Marie-Tooth disease].
Hereditary motor and sensory type 1A neuropathy (known as Charcot-Marie-Tooth disease) is a disease of peripheral nerves characterized by symptoms of progressive polyneuropathy with preferential damage of distal extremity muscles. Damage to the cardiovascular system is extremely rare and heterogenous in this pathology. This disease is not included in the list of indications for interventional antiarrhythmic aid. We could not find in available literature a clinical description of the development of sinus node dysfunction associated with this pathology. The present clinical report presents a case of detection and successful treatment of a damage to the cardiovascular system that manifested itself as sinus node dysfunction/sick sinus syndrome in the tachy-brady variant. A combination treatment approach using radiofrequency catheter ablation, implantation of a permanent pacemaker, and antiarrhythmic therapy associated with drug and non-drug treatment of motor sensory neuropathy resulted in recovery and long-term maintenance of sinus rhythm as well as in beneficial changes in the patient's neurological status.
Identification of rare heterozygous linkage R965C-R1309H mutations in the pore-forming region of SCN5A gene associated with complex arrhythmia.
We examined the genetic background of a Chinese Han family in which some members presented with complex arrhythmias including sick sinus syndrome, progressive conduction block, atrial fibrillation, atrial standstill and Brugada syndrome. The possible underlying mechanism associated with the genetic mutation was explored. Targeted capture sequencing was conducted in the probands in the coding and splicing regions of genes implicated in inherited arrhythmias. Stable cell lines overexpressing wild type (WT) or mutant SCN5A were generated in HEK293T cells. Whole-cell recording was performed to evaluate the functional changes in sodium channels. The rare heterozygous linkage mutations, SCN5A R965C and R1309H, were found in these patients with complex familial arrhythmias. Compared to WT, R965C or R1309H, the peak current of sodium channel was dramatically reduced in HEK293T cell with linkage R965C-R1309H mutation when testing potentials ranging from -45 to 15 mV. Notably, the maximum peak current of sodium channels with R1309H and linkage R965C-R1309H displayed significant decreases of 31.5% and 73.34%, respectively, compared to WT. Additionally, compared to R965C or R1309H alone, the linkage mutation R965C-R1309H demonstrated not only a more obvious depolarisation-shifted activation and hyperpolarisation-shifted inactivation, but also a more significant alteration in the time constant, V1/2 and the slope factor of activation and inactivation. The linkage mutation SCN5A R965C-R1309H led to a more dramatically reduced current density, as well as more significant depolarisation-shifted activation and hyperpolarisation-shifted inactivation in sodium channels than R965C or R1309H alone, which potentially explain this complex familial arrhythmia syndrome.
Publicações recentes
[Polymorphisms of 2B-adrenergic receptor and endothelial NO-Synthase genes in genesis of the hereditary sick sinus node syndrome].
[Polymorphism of connexin 40 gene-- a novel genetic marker of the sick sinus node syndrome].
[Ser49gly polymorphism as predictor of development of hereditary sick sinus node syndrome].
📚 EuropePMCmostrando 16
Adult late-onset limb-girdle muscular dystrophy R1/2A complicated by parathyroid adenoma and sick sinus syndrome: a case report and literature review.
BMC musculoskeletal disordersSystematic analysis of SCN5A variants associated with inherited cardiac diseases.
Heart rhythmIncidence and predictors of pacemaker implantation at follow-up after reversible high-degree sinus node dysfunction or atrioventricular block.
Pacing and clinical electrophysiology : PACE[Clinical case of the cardiovascular system involvement in a patient with Charcot-Marie-Tooth disease].
KardiologiiaIdentification of rare heterozygous linkage R965C-R1309H mutations in the pore-forming region of SCN5A gene associated with complex arrhythmia.
Molecular genetics & genomic medicineDisease-associated HCN4 V759I variant is not sufficient to impair cardiac pacemaking.
Pflugers Archiv : European journal of physiologyPrevalence, Incidence, and Impact on Mortality of Conduction System Disease in Transthyretin Cardiac Amyloidosis.
The American journal of cardiology[Sinus node dysfunction, Brugada syndrome and long QT syndrome affecting the same patient : when genetics can't make head or tail of it].
Revue medicale suisseGeneration of two iPSC lines (FAMRCi004-A and FAMRCi004-B) from patient with familial progressive cardiac conduction disorder carrying genetic variant DSP p.His1684Arg.
Stem cell researchSevere congenital RYR1-associated myopathy complicated with atrial tachycardia and sinus node dysfunction: a case report.
Italian journal of pediatricsGenetic mutation of familial dilated cardiomyopathy based on next‑generation semiconductor sequencing.
Molecular medicine reportsClinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy.
JACC. Clinical electrophysiologyGenotype-phenotype dilemma in a case of sudden cardiac death with the E1053K mutation and a deletion in the SCN5A gene.
Forensic science internationalVariants in the SCN5A Promoter Associated With Various Arrhythmia Phenotypes.
Journal of the American Heart AssociationGenetic analysis of cardiac SCN5A Gene in Iranian patients with hereditary cardiac arrhythmias.
Anatolian journal of cardiologySide effects of ticagrelor: Sinus node dysfunction with ventricular pause.
International journal of cardiologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome do nó sinusal doente hereditária.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome do nó sinusal doente hereditária
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Systematic analysis of SCN5A variants associated with inherited cardiac diseases.
- Adult late-onset limb-girdle muscular dystrophy R1/2A complicated by parathyroid adenoma and sick sinus syndrome: a case report and literature review.
- Incidence and predictors of pacemaker implantation at follow-up after reversible high-degree sinus node dysfunction or atrioventricular block.
- [Clinical case of the cardiovascular system involvement in a patient with Charcot-Marie-Tooth disease].
- Identification of rare heterozygous linkage R965C-R1309H mutations in the pore-forming region of SCN5A gene associated with complex arrhythmia.
- [Polymorphisms of 2B-adrenergic receptor and endothelial NO-Synthase genes in genesis of the hereditary sick sinus node syndrome].
- [Polymorphism of connexin 40 gene-- a novel genetic marker of the sick sinus node syndrome].
- [Ser49gly polymorphism as predictor of development of hereditary sick sinus node syndrome].
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:166282(Orphanet)
- MONDO:0012061(MONDO)
- GARD:13663(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q56014648(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome do nó sinusal doente hereditária
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata