Síndrome epiléptica genética caracterizada pela ocorrência de crises afebris repetidas em bebês saudáveis, entre o terceiro e o oitavo mês de vida.
Introdução
O que você precisa saber de cara
Síndrome epiléptica genética caracterizada pela ocorrência de crises afebris repetidas em bebês saudáveis, entre o terceiro e o oitavo mês de vida.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 13 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 49 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
5 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant.
As a component of the outer core of AMPAR complex, may be involved in synaptic transmission in the central nervous system. In hippocampal neurons, in presynaptic terminals, plays an important role in the final steps of neurotransmitter release, possibly by regulating Ca(2+)-sensing. In the cerebellum, may inhibit SNARE complex formation and down-regulate short-term facilitation
Cell membranePresynaptic cell membraneSynapseCell projection, axonCytoplasmic vesicle, secretory vesicle, synaptic vesicle membranePostsynaptic density membraneCell projection, dendritic spine
Episodic kinesigenic dyskinesia 1
An autosomal dominant form of paroxysmal kinesigenic dyskinesia, a neurologic condition characterized by recurrent and brief attacks of abnormal involuntary movements, triggered by sudden voluntary movement. These attacks usually have onset during childhood or early adulthood and can involve dystonic postures, chorea, or athetosis.
Pore-forming subunit of the voltage-gated potassium (Kv) M-channel which is responsible for the M-current, a key controller of neuronal excitability (PubMed:16319223, PubMed:27564677, PubMed:28793216, PubMed:9872318). M-channel is composed of pore-forming subunits KCNQ2 and KCNQ3 assembled as heterotetramers (PubMed:14534157, PubMed:16319223, PubMed:27564677, PubMed:9872318). The native M-current has a slowly activating and deactivating potassium conductance which plays a critical role in determ
Cell membrane
Seizures, benign familial neonatal 2
A disorder characterized by clusters of seizures occurring in the first days of life. Most patients have spontaneous remission by 12 months of age and show normal psychomotor development. The disorder is distinguished from benign familial infantile seizures by an earlier age at onset.
Mediates the voltage-dependent sodium ion permeability of excitable membranes. Assuming opened or closed conformations in response to the voltage difference across the membrane, the protein forms a sodium-selective channel through which Na(+) ions may pass in accordance with their electrochemical gradient (PubMed:1325650, PubMed:17021166, PubMed:28256214, PubMed:29844171). Implicated in the regulation of hippocampal replay occurring within sharp wave ripples (SPW-R) important for memory (By simi
Cell membrane
Seizures, benign familial infantile, 3
A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS3 inheritance is autosomal dominant.
Pore-forming subunit of the voltage-gated potassium (Kv) M-channel which is responsible for the M-current, a key controller of neuronal excitability (PubMed:24277843, PubMed:28793216, PubMed:9836639). M-channel is composed of pore-forming subunits KCNQ2 and KCNQ3 assembled as heterotetramers (PubMed:10781098, PubMed:14534157, PubMed:32884139, PubMed:37857637, PubMed:9836639). The native M-current has a slowly activating and deactivating potassium conductance which plays a critical role in determ
Cell membrane
Seizures, benign familial neonatal 1
A disorder characterized by clusters of seizures occurring in the first days of life. Most patients have spontaneous remission by 12 months of age and show normal psychomotor development. Some rare cases manifest an atypical severe phenotype associated with epileptic encephalopathy and psychomotor retardation. The disorder is distinguished from benign familial infantile seizures by an earlier age at onset. In some patients, neonatal convulsions are followed later in life by myokymia, a benign condition characterized by spontaneous involuntary contractions of skeletal muscles fiber groups that can be observed as vermiform movement of the overlying skin. Electromyography typically shows continuous motor unit activity with spontaneous oligo- and multiplet-discharges of high intraburst frequency (myokymic discharges). Some patients may have isolated myokymia.
Pore-forming subunit of a voltage-gated sodium channel complex assuming opened or closed conformations in response to the voltage difference across membranes and through which sodium ions selectively pass along their electrochemical gradient (PubMed:24874546, PubMed:25239001, PubMed:25725044, PubMed:26900580, PubMed:29726066, PubMed:33245860, PubMed:36696443, PubMed:36823201). Contributes to neuronal excitability by regulating action potential threshold and propagation (PubMed:24874546, PubMed:2
Cell membraneCell projection, axonCytoplasmic vesicleCell projection, podosome
Cognitive impairment with or without cerebellar ataxia
A disorder characterized by markedly delayed cognitive and motor development, attention deficit disorder, and cerebellar ataxia. Features include bilateral esophoria, strabismatic amblyopia, unsustained gaze evoked nystagmus on horizontal gaze, ataxic gait, dysmetria in the upper limbs and dysarthria, with normal strength, tone, and reflexes.
Variantes genéticas (ClinVar)
2.147 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
4 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Epilepsia infantil auto-limitada
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Clinical and neuroimaging features of PRRT2-related epilepsy in adult patients.
While proline-rich transmembrane protein 2 (PRRT2) variants have been reported in association with self-limited infantile epilepsy (SeLIE), their relevance to adult epilepsy remains largely uncharacterized. Therefore, we investigated the prevalence of PRRT2 variants in an adult epilepsy cohort to broaden their phenotypic spectrum. In a cohort of adult patients with epilepsy who underwent whole-exome sequencing (WES), individuals harboring PRRT2 variants were identified and selected for further analysis. Amino acid conservation among species, combined with functional verification of mutant proteins, was performed to assess the pathogenicity of novel PRRT2 variants. Furthermore, electroencephalography (EEG) and functional magnetic resonance imaging (fMRI) were conducted in patients carrying PRRT2 variants. Two novel missense variants (c.643C > A/ p.P215T and c.932G > A/ p.R311Q) were demonstrated to impair protein expression, indicating a likely haploinsufficiency mechanism. Nine cases were identified carrying PRRT2 variants, including the c.649dupC hotspot variant and two missense variants. The age of seizure onset was predominantly during infancy or adolescence, with epilepsy persisting or relapsing into adulthood. Drug-resistant epilepsy was observed in two cases, whereas others were primarily controlled through sodium channel blockers. Unlike previously reported normal interictal EEG findings in PRRT2-related disorders, epileptiform discharges were recorded in the majority of patients. fMRI revealed reduced thalamocortical connectivity, indicating disrupted brain network integration. This study expands the genotypic and phenotypic spectrum of PRRT2-related disorders, links its variants to epileptiform discharges, and reveals impaired network integration in PRRT2-associated epilepsy.
Distinctive genetic architecture of infantile epileptic spasms syndrome compared to self-limited infantile epilepsy by trios whole-exome sequencing.
Infantile epileptic spasms syndrome (IESS) and self-limited infantile epilepsy (SeLIE) are both genetically heterogeneous disorders during infancy with distinct prognoses. To better define the genetic spectrum of IESS, we performed a comparative genetic analysis using SeLIE cases as a reference group. We performed whole-exome sequencing in a Chinese cohort comprising 54 parent-offspring trios with IESS and 37 trios or quartets with SeLIE. Causal pathogenic or likely pathogenic variants were identified in 61% (56/92) of patients, distributed across 20 different genes. Among these, 10 (29%) variants were novel. A definitive genetic diagnosis was achieved in 37% (20/54) of IESS patients, involving 17 distinct genes, indicating a high degree of genetic heterogeneity. In contrast, SeLIE patients had a much higher diagnostic yield of 95% (36/38), with variants concentrated in only four genes. By comparing de novo mutations in protein-coding regions between the IESS and SeLIE cohorts, we identified both shared and potentially novel variants. Notably, there was a trend toward an increased burden of de novo loss-of-function variants in IESS compared to SeLIE. Among IESS patients, 51.9% (28/54) responded to initial hormonal therapy, while 38.9% (21/54) failed to respond. These integrative genomic analyses provide new insights into the pathogenesis of IESS, underscoring a more complex and heterogeneous genetic landscape in IESS compared to SeLIE. This study investigated the genetic architecture of two infantile epilepsy syndromes: IESS and SeLIE. We demonstrated that IESS has a more heterogeneous genetic background, whereas SeLIE is associated with more specific variants. These findings provide novel insights into the genetic underpinnings of IESS.
Multiple phenotypic traits including developmental impairment in a Chinese family with infantile convulsion and choreoathetosis syndrome: a case study expanding the clinical spectrum of prrt2-related syndrome.
Pathogenic heterozygous variants in the gene encoding proline-rich transmembrane protein 2 (PRRT2) have been recently identified as the major cause of familial infantile convulsion and choreoathetosis syndrome (OMIM#602,066), a spectrum of autosomal dominant paroxysmal neurological disorders, including self-limited infantile epilepsy (SeLIE) and infantile convulsion that can be isolated (IC) or associated with paroxysmal kinesigenic dyskinesia (PKD/IC). Incomplete penetrance of PRRT2 variants and variable phenotypes without developmental impairment have been widely reported in previous studies of this syndrome, but no studies to date have documented global development delay (GDD) with growth retardation (GR) occurred in a family with multiple phenotypes of this syndrome. Here, using family-based whole-exome sequencing, we identified a pathogenic heterozygous PRRT2 variant (NM_145239.3: c.718C > T, p.Arg240*) in a 3-generation Chinese family of infantile convulsion and choreoathetosis syndrome. The variant was detected in five family members, of which two (pedigree III.1 and III.3) were diagnosed with PKD/IC, one (pedigree III.2) presented uncontrolled generalized/focal seizures with GDD and GR; the GR of this patient was aggravated with the progression of the epileptic condition; she was then diagnosed with IC and developmental impairment, one (pedigree II.2) was diagnosed with SeLIE, and one (pedigree II.3) was phenotypically unaffected and recognized as an obligate carrier. In conclusion, we reported a PRRT2-related syndrome family harboring multiple phenotypic features, including uncontrolled seizures with developmental impairment, which may potentially expand PRRT2-related clinical spectrum. Moreover, our findings suggest that children with PRRT2-related seizures/convulsions, especially those who suffer from uncontrolled multiple seizure types, should be aware of potential risks of having developmental impairment aggravation and need timely and effective antiepileptic medications.
PRRT 2-Related Epilepsy: From Self-Limited Infantile Epilepsy to Atypical Epilepsy Phenotypes.
Pathogenic variants in the PRRT2 gene cause self-limited infantile epilepsy (SeLIE). Recently, atypical epilepsy phenotypes have been described. We explore the phenotypic spectrum of PRRT2-related epilepsy through international collaboration. All children with epilepsy and either a pathogenic PRRT2 variant or 16p11.2 microdeletion encompassing the PRRT2 gene were included in this retrospective study. Details related to the epilepsy, comorbidities, genetic results, EEG/neuroimaging findings, and treatments are summarized. Forty children were identified, and 24 were male (n = 24/40, 60%). The median age at seizure onset was 5 months (IQR 4, 6) (range 2-150 months). Bilateral tonic-clonic (BTC) (n = 22/40, 55%) and focal motor seizures with impaired awareness evolving to BTC seizures (n = 9/40, 23%) were most common. Thirty-six children (n = 36/40, 90%) had pathogenic PRRT2 variants; 3 were homozygous, and 33 were heterozygous. Four children (n = 4/40, 10%) had 16p11.2 microdeletion. SeLIE was most common, diagnosed in 32 children with heterozygous PRRT2 variants (n = 32/33, 97%), 3 children with homozygous PRRT2 variants (n = 3/3, 100%), and 3 children with 16p11.2 microdeletion (n = 3/4, 75%). Atypical phenotypes were observed in 3 children with heterozygous PRRT2 variants; 1 child evolved from SeLIE to infantile epileptic spasms, another developed spike-wave activation in sleep, and 1 developed focal epilepsy in adolescence. Medically refractory genetic generalized epilepsy and intellectual disability were diagnosed in 1 child with a whole-gene PRRT2 deletion and 16p11.2 microdeletion. All children with homozygous PRRT2 variants had SeLIE with movement disorders. Thirty-seven children (n = 37/40, 93%) were treated with antiseizure medications, and sodium channel blockers were effective in most (20/27 responded, 74%). The median age at seizure freedom was 9 months (IQR 3, 10) (range 3-168 months). Pathogenic PRRT2 variants are commonly associated with SeLIE. However, additional epilepsy phenotypes may be observed with heterozygous PRRT2 variants. In individuals with heterozygous PRRT2 variants, corresponding chromosomal microarray may be helpful to assess for concomitant 16p11.2 microdeletion, given the phenotypic overlap between the 2 conditions. Collection of additional cases is needed, however, to better understand the spectrum of epilepsy phenotypes associated with 16p11.2 microdeletion encompassing the PRRT2 gene and homozygous and compound heterozygous PRRT2 variants. Currently, precise genotype-phenotype relationships are lacking.
What is the right choice? Is the answer sodium channel blockers?
Self-limited infantile epilepsy (SeLIE) is one of the most common epileptic syndromes encountered in infancy. The rapid control of seizures and determining the etiology will help the clinician and the family. Care providers need to be aware of the implications, etiology, and management. In our study, the aim is to evaluate the characteristics and response to treatment in children with SeLIE and to raise awareness about early diagnosis and treatment options. A total of 72 infants and children diagnosed with SeLIE were retrospectively analyzed to evaluate their clinical and prognostic data. The average age at the time of diagnosis of patients was 7.6 ± 4 months, and the onset of seizures was 7 ± 3.7 months, and 54.1% were boys in the study group. Of the patients with normal neurodevelopmental steps, 38 (52.7%) had focal elementary motor seizures, 17 (23.6%) had focal impaired consciousness seizures with observable manifestation, 16 (22.2%) had bilateral tonic-clonic seizures of unknown onset, and 1 had focal to bilateral tonic-clonic seizure initially. The most common anti-seizure medication chosen as the first-line treatment was levetiracetam in our study cohort, but the highest seizure-free rate was achieved by initiating carbamazepine. If focal seizures occur in clusters in an afebrile infant with normal neuromotor development and normal neuroimaging, SeLIE should be considered as a possible diagnosis. Sodium channel blockers, with shown high treatment success, should be considered as the first-line medication for favorable outcomes in SeLIE.
Publicações recentes
Clinical and neuroimaging features of PRRT2-related epilepsy in adult patients.
Distinctive genetic architecture of infantile epileptic spasms syndrome compared to self-limited infantile epilepsy by trios whole-exome sequencing.
Multiple phenotypic traits including developmental impairment in a Chinese family with infantile convulsion and choreoathetosis syndrome: a case study expanding the clinical spectrum of prrt2-related syndrome.
What is the right choice? Is the answer sodium channel blockers?
PRRT 2-Related Epilepsy: From Self-Limited Infantile Epilepsy to Atypical Epilepsy Phenotypes.
📚 EuropePMC7 artigos no totalmostrando 17
Clinical and neuroimaging features of PRRT2-related epilepsy in adult patients.
Epilepsy & behavior : E&BDistinctive genetic architecture of infantile epileptic spasms syndrome compared to self-limited infantile epilepsy by trios whole-exome sequencing.
Epilepsia openMultiple phenotypic traits including developmental impairment in a Chinese family with infantile convulsion and choreoathetosis syndrome: a case study expanding the clinical spectrum of prrt2-related syndrome.
BMC pediatricsWhat is the right choice? Is the answer sodium channel blockers?
Epileptic disorders : international epilepsy journal with videotapePRRT 2-Related Epilepsy: From Self-Limited Infantile Epilepsy to Atypical Epilepsy Phenotypes.
Neurology. Genetics[Contribution of the Ibero-American Academy of Neuropediatrics to the knowledge of self-limited epilepsy in infants].
MedicinaVoltage-gated sodium channel epilepsies in a tertiary care center: Phenotypic spectrum with correlation to predicted functional effects.
Epilepsy & behavior : E&BSCN8A self-limited infantile epilepsy: Does epilepsy resolve?
EpilepsiaClinical and genetic analysis of 18 patients with KCNQ2 mutations from South China.
The Turkish journal of pediatricsEpilepsies with onset during the first year of life: A prospective study on syndromes, etiologies, and outcomes.
Epilepsia openBenign convulsions with mild gastroenteritis in children: An emerging acute symptomatic seizures.
Pediatric discoveryBi-allelic PRRT2 variants may predispose to Self-limited Familial Infantile Epilepsy.
European journal of human genetics : EJHGLevetiracetam may be an unsuitable choice for patients with PRRT2-associated self-limited infantile epilepsy.
BMC pediatricsPeri-ictal EEG in infants with PRRT2-related self-limited infantile epilepsy.
Epileptic disorders : international epilepsy journal with videotapePRRT2-positive self-limited infantile epilepsy: Initial seizure characteristics and response to sodium channel blockers.
Epilepsia openProbability of Remission of the Main Epileptic Syndromes in Childhood.
Journal of child neurologyUncommon epileptic syndromes in children: a review.
SeizureAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Epilepsia infantil auto-limitada.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Epilepsia infantil auto-limitada
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Clinical and neuroimaging features of PRRT2-related epilepsy in adult patients.
- Distinctive genetic architecture of infantile epileptic spasms syndrome compared to self-limited infantile epilepsy by trios whole-exome sequencing.
- Multiple phenotypic traits including developmental impairment in a Chinese family with infantile convulsion and choreoathetosis syndrome: a case study expanding the clinical spectrum of prrt2-related syndrome.
- PRRT 2-Related Epilepsy: From Self-Limited Infantile Epilepsy to Atypical Epilepsy Phenotypes.
- What is the right choice? Is the answer sodium channel blockers?
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:306(Orphanet)
- MONDO:0017615(MONDO)
- Epilepsia(PCDT · Ministério da Saúde)
- GARD:857(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q4887955(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
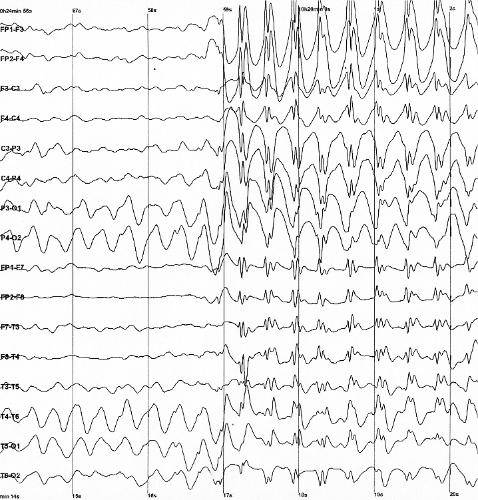
Epilepsia infantil auto-limitada
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub