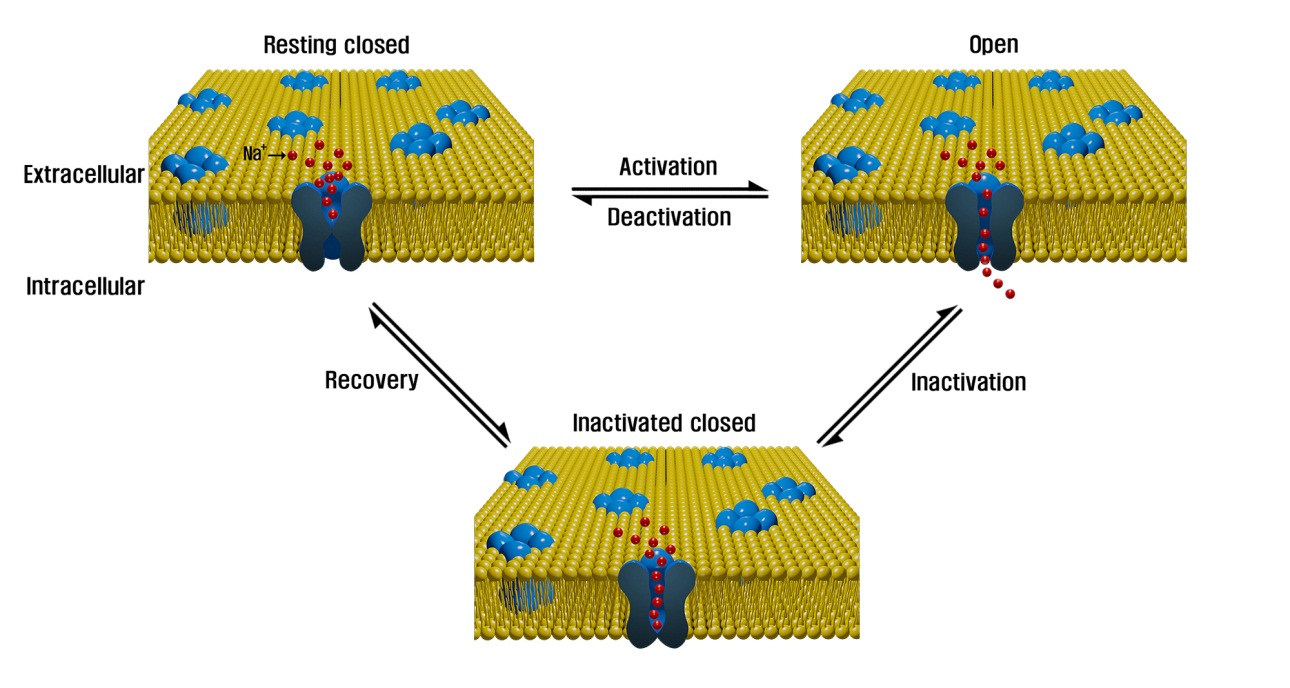
A paralisia periódica hipercalêmica é um distúrbio autossômico dominante hereditário que afeta os canais de sódio nas células musculares e a capacidade de regular os níveis de potássio no sangue. É caracterizada por hiperexcitabilidade ou fraqueza muscular que, exacerbada pelo potássio, calor ou frio, pode levar a tremores incontroláveis seguidos de paralisia. O início geralmente ocorre na primeira infância, mas ainda ocorre em adultos.
Introdução
O que você precisa saber de cara
Visão geral
A paralisia periódica normocalêmica é uma doença rara caracterizada por episódios de fraqueza muscular ou paralisia que ocorrem sem alteração nos níveis de potássio no sangue (normocalemia). A condição faz parte de um grupo de doenças chamadas paralisias periódicas, que afetam a função dos músculos esqueléticos. Ainda não há dados precisos sobre sua prevalência na população.[1]
Sinais e sintomas
Os principais sintomas são episódios súbitos de fraqueza muscular ou paralisia, que podem afetar braços, pernas e tronco. Diferente de outras formas de paralisia periódica, os níveis de potássio no sangue permanecem normais durante as crises. A idade de início dos sintomas não está claramente definida na literatura disponível.[1]
Causas genéticas
A paralisia periódica normocalêmica é uma condição genética, mas os genes específicos envolvidos ainda não foram identificados de forma conclusiva. A doença está catalogada no banco de dados OMIM (OMIM:170600) e na ontologia MONDO (MONDO:0008225), indicando seu reconhecimento como entidade clínica distinta.[1][2][4]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos episódios de paralisia e na confirmação de que os níveis de potássio permanecem normais durante as crises. Exames genéticos podem ser utilizados para investigar mutações associadas, embora os genes específicos ainda não estejam totalmente definidos. Não há informações disponíveis sobre testes genéticos específicos ou variantes no ClinVar para esta condição.[1][2][4][5]
Tratamento e manejo
O manejo da paralisia periódica normocalêmica foca no controle dos episódios e na prevenção de crises. Não há um protocolo padronizado de tratamento, e as abordagens são baseadas em relatos da literatura e experiência clínica. É importante que o acompanhamento seja feito por uma equipe médica especializada.[1][2][5]
Tratamentos citados na literatura
A literatura científica (fonte: PubTator3) menciona associações entre a paralisia periódica normocalêmica e algumas substâncias, com base no número de publicações. Estas são associações mineradas de artigos científicos e não representam recomendações de tratamento. As substâncias mais citadas são: Potássio (52 publicações), Acetazolamida (26 publicações), Sódio (25 publicações), Diclorfenamida (16 publicações), Cálcio (7 publicações), Cloreto de Potássio (7 publicações), Carboidratos (5 publicações), Cloretos (4 publicações), Albuterol (4 publicações) e Bumetanida (4 publicações).[5]
Prognóstico e qualidade de vida
Não há informações específicas na literatura disponível sobre o prognóstico de longo prazo ou impacto na qualidade de vida para a paralisia periódica normocalêmica. O acompanhamento médico regular é essencial para monitorar a evolução da condição.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
A paralisia periódica hipercalêmica é um distúrbio autossômico dominante hereditário que afeta os canais de sódio nas células musculares e a capacidade de regular os níveis de potássio no sangue. É caracterizada por hiperexcitabilidade ou fraqueza muscular que, exacerbada pelo potássio, calor ou frio, pode levar a tremores incontroláveis seguidos de paralisia. O início geralmente ocorre na primeira infância, mas ainda ocorre em adultos.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A paralisia periódica normocalêmica é uma doença rara caracterizada por episódios de fraqueza muscular ou paralisia que ocorrem sem alteração nos níveis de potássio no sangue (normocalemia). A condição faz parte de um grupo de doenças chamadas paralisias periódicas, que afetam a função dos músculos esqueléticos. Ainda não há dados precisos sobre sua prevalência na população.[1]
Sinais e sintomas
Os principais sintomas são episódios súbitos de fraqueza muscular ou paralisia, que podem afetar braços, pernas e tronco. Diferente de outras formas de paralisia periódica, os níveis de potássio no sangue permanecem normais durante as crises. A idade de início dos sintomas não está claramente definida na literatura disponível.[1]
Causas genéticas
A paralisia periódica normocalêmica é uma condição genética, mas os genes específicos envolvidos ainda não foram identificados de forma conclusiva. A doença está catalogada no banco de dados OMIM (OMIM:170600) e na ontologia MONDO (MONDO:0008225), indicando seu reconhecimento como entidade clínica distinta.[1][2][4]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos episódios de paralisia e na confirmação de que os níveis de potássio permanecem normais durante as crises. Exames genéticos podem ser utilizados para investigar mutações associadas, embora os genes específicos ainda não estejam totalmente definidos. Não há informações disponíveis sobre testes genéticos específicos ou variantes no ClinVar para esta condição.[1][2][4][5]
Tratamento e manejo
O manejo da paralisia periódica normocalêmica foca no controle dos episódios e na prevenção de crises. Não há um protocolo padronizado de tratamento, e as abordagens são baseadas em relatos da literatura e experiência clínica. É importante que o acompanhamento seja feito por uma equipe médica especializada.[1][2][5]
Tratamentos citados na literatura
A literatura científica (fonte: PubTator3) menciona associações entre a paralisia periódica normocalêmica e algumas substâncias, com base no número de publicações. Estas são associações mineradas de artigos científicos e não representam recomendações de tratamento. As substâncias mais citadas são: Potássio (52 publicações), Acetazolamida (26 publicações), Sódio (25 publicações), Diclorfenamida (16 publicações), Cálcio (7 publicações), Cloreto de Potássio (7 publicações), Carboidratos (5 publicações), Cloretos (4 publicações), Albuterol (4 publicações) e Bumetanida (4 publicações).[5]
Prognóstico e qualidade de vida
Não há informações específicas na literatura disponível sobre o prognóstico de longo prazo ou impacto na qualidade de vida para a paralisia periódica normocalêmica. O acompanhamento médico regular é essencial para monitorar a evolução da condição.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Paralisia periódica normocalêmica
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Appeared inexplicable disorders of consciousness after general anesthesia tracheal tube drawing in endoscopic tympanoplasty.
Disorders of consciousness (DOC) are neurocognitive disorders related to sharp fluctuations of attention and consciousness, while DOC is characterized by significant interindividual differences, rapid development, and a higher lethal rate. A 53-year-old female patient underwent general anesthesia with tracheal intubation in otoendoscopic tympanoplasty. The patient suddenly appeared moderate DOC after tracheal tube removal with K+ 3.6 (3.5-5.3 mmol/L). Based on the ancillary testing and routine laboratory workup, the possible causes of DOC, such as general anesthesia drugs and cardio cerebral events, were temporarily excluded. DOC was reversed by intravenous administration of KCl 1 g, with K+ 3.78 mmol/L. On one day after surgery, the patient occurred suddenly DOC again after intravenous guttae of 5% glucose 1000 ml, K+ 3.87 mmol/L, possibly because of her recurrent hypokalemic paralysis (HP) of past medical history. The patient's consciousness gradually improved after effective KCl supplementation therapy. DOC caused by periodic paralysis (PP) has not been reported, we speculate that hypoactive DOC is closely correlated with normokalemic periodic paralysis (NormoPP) in this case.
The clinical and genetic heterogeneity analysis of five families with primary periodic paralysis.
To explore the clinical and genetic characteristics of five families with primary periodic paralysis (PPP). We reviewed clinical manifestations, laboratory results, electrocardiogram, electromyography, muscle biopsy, and genetic analysis from five families with PPP. Five families with PPP included: hypokalemic periodic paralysis type 1 (HypoPP1, CACNA1S, 1/5), hypokalemic periodic paralysis type 2 (HypoPP2, SCN4A, 2/5), normokalemic periodic paralysis (NormoPP, SCN4A, 1/5), and Andersen-Tawil syndrome (ATS, KCNJ2, 1/5). The basic clinical manifestations of five families were consistent with PPP, presenting with paroxysmal muscle weakness, with or without abnormal serum potassium. ATS was accompanied by ventricular arrhythmias, and skeletal and craniofacial anomalies, developing with a permanent fixed myopathy later. The electromyography showed diffuse myopathic discharge, and muscle biopsy showed tubular aggregates. Genetic testing revealed five families with PPP carried CACNA1S (R1242S), SCN4A (R675Q, T704M), and KCNJ2 (R218Q) respectively. The novel heterozygous R1242S mutation in CACNA1S caused a conformational change in the protein structure, and the amino acid of this mutation site was highly conserved among different species. SCN4A mutations led to two phenotypes of HypoPP2 and NormoPP. PPPs are autosomal dominant disorders of ion channel dysfunction characterized by episodic flaccid muscle weakness secondary to abnormal sarcolemmal excitability. PPPs are caused by mutations in skeletal muscle calcium channel CaV1.1 gene (CACNA1S), sodium channel NaV1.4 gene (SCN4A), and potassium channels Kir2.1, Kir3.4 genes (KCNJ2, KCNJ5), including HypoPP1, HypoPP2, NormoPP, HyperPP, and ATS, which have significant clinical and genetic heterogeneity. Diagnosis is based on the characteristic clinical presentation then confirmed by genetic testing.
Identification of a SCN4A mutation in a large Chinese family with atypical normokalemic periodic paralysis using whole-exome sequencing.
Normokalemic periodic paralysis (NormoKPP) of skeletal muscle is an autosomal dominant disorder caused by mutations in the gene encoding voltage-gated sodium channel protein type 4 subunit alpha (SCN4A), which leads to ion channel dysfunction. Little is known about the relationship between genotype and the clinical symptoms of NormoKPP. The present study aimed to evaluate the genetic variation in a large Chinese family with NormoKPP. The patients in this pedigree did not respond to saline treatment, but calcium gluconate treatment was effective. We performed a series of clinical examinations and genetic analyses, using whole-exome and Sanger sequencing, to examine the mutation status of SCN4A in a Chinese family segregating for NormoKPP. Whole-exome sequencing revealed a c.2111C>T substitution in SCN4A in most of the affected family members. This mutation results in the amino acid substitution p.T704M. These results support a causative role of this mutation in SCN4A in NormoKPP, and provide information about the relationship between genotype and atypical clinical symptoms.
Skeletal muscle CaV1.1 channelopathies.
CaV1.1 is specifically expressed in skeletal muscle where it functions as voltage sensor of skeletal muscle excitation-contraction (EC) coupling independently of its functions as L-type calcium channel. Consequently, all known CaV1.1-related diseases are muscle diseases and the molecular and cellular disease mechanisms relate to the dual functions of CaV1.1 in this tissue. To date, four types of muscle diseases are known that can be linked to mutations in the CACNA1S gene or to splicing defects. These are hypo- and normokalemic periodic paralysis, malignant hyperthermia susceptibility, CaV1.1-related myopathies, and myotonic dystrophy type 1. In addition, the CaV1.1 function in EC coupling is perturbed in Native American myopathy, arising from mutations in the CaV1.1-associated protein STAC3. Here, we first address general considerations concerning the possible roles of CaV1.1 in disease and then discuss the state of the art regarding the pathophysiology of the CaV1.1-related skeletal muscle diseases with an emphasis on molecular disease mechanisms.
SCN4A p.R675Q Mutation Leading to Normokalemic Periodic Paralysis: A Family Report and Literature Review.
Objective: To investigate the clinical features, skeletal muscle imaging, and muscle pathological characteristics of normokalemic periodic paralysis (NormoKPP) caused by mutation of SCN4A gene p.R675Q. Methods: The clinical data, skeletal muscle imaging, pathological data, and gene test results of a family with NormoKPP were collected in detail in October 2018. The previous literature was reviewed and used for comparative analysis. Results: The proband was a 28-year-old male with paroxysmal weakness of both lower limbs for 14 years. Limb weakness was mainly manifested in the proximal extremities of both lower limbs, which occurred two to three times a year. The muscle weakness of each attack lasted for 1-2 weeks and gradually recovered. The blood potassium levels were normal. The abnormal signals of the posterior thigh muscle group and the medial calf muscle group could be seen on the magnetic resonance imaging (MRI) of the skeletal muscle, and the target-fiber could be seen in some muscle fibers in muscle pathology. The father of the proband and his brother had the same symptoms. In the same family, 10 people received genetic testing. The results showed that five had a mutation of SCN4A gene p.R675Q. The mutation gene came from the father of the proband. Conclusion: NormoKPP is a clinically rare form of sodium ion channel disease. The clinical manifestations, skeletal muscle imaging, and pathological changes are different from the common hypokalemic periodic paralysis. SCN4A gene detection is an important means for the diagnosis of NormoKPP.
Publicações recentes
Lifestyle and dietary measures in Periodic Paralyses.
Thyrotoxic Paralysis in a Hispanic Woman: An Unusual Presentation of a Neurological Emergency.
Appeared inexplicable disorders of consciousness after general anesthesia tracheal tube drawing in endoscopic tympanoplasty.
The clinical and genetic heterogeneity analysis of five families with primary periodic paralysis.
Identification of a SCN4A mutation in a large Chinese family with atypical normokalemic periodic paralysis using whole-exome sequencing.
📖 Revisão📚 EuropePMC24 artigos no totalmostrando 11
Appeared inexplicable disorders of consciousness after general anesthesia tracheal tube drawing in endoscopic tympanoplasty.
IbrainThe clinical and genetic heterogeneity analysis of five families with primary periodic paralysis.
Channels (Austin, Tex.)Identification of a SCN4A mutation in a large Chinese family with atypical normokalemic periodic paralysis using whole-exome sequencing.
The Journal of international medical researchSkeletal muscle CaV1.1 channelopathies.
Pflugers Archiv : European journal of physiologySCN4A p.R675Q Mutation Leading to Normokalemic Periodic Paralysis: A Family Report and Literature Review.
Frontiers in neurology[Analysis of SCN4A gene variation in a Chinese pedigree affected with skeletal muscle sodium channelopathies].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsIncreased KCNJ18 promoter activity as a mechanism in atypical normokalemic periodic paralysis.
Neurology. GeneticsFamilial Normokalemic Periodic Paralysis Associated With Mutation in the SCN4A p.M1592V.
Frontiers in neurologySuccessful treatment of normokalemic periodic paralysis with hydrochlorothiazide.
Brain & developmentDe novo Mutation in CACNA1S Gene in a 20-Year-Old Man Diagnosed with Metabolic Myopathy.
Archives of Iranian medicineMutations of SCN4A gene cause different diseases: 2 case reports and literature review.
Channels (Austin, Tex.)Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Paralisia periódica normocalêmica.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Paralisia periódica normocalêmica
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Appeared inexplicable disorders of consciousness after general anesthesia tracheal tube drawing in endoscopic tympanoplasty.
- The clinical and genetic heterogeneity analysis of five families with primary periodic paralysis.
- Identification of a SCN4A mutation in a large Chinese family with atypical normokalemic periodic paralysis using whole-exome sequencing.
- Skeletal muscle CaV1.1 channelopathies.
- SCN4A p.R675Q Mutation Leading to Normokalemic Periodic Paralysis: A Family Report and Literature Review.
- Lifestyle and dietary measures in Periodic Paralyses.
- Thyrotoxic Paralysis in a Hispanic Woman: An Unusual Presentation of a Neurological Emergency.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:680(Orphanet)
- MONDO:0008225(MONDO)
- GARD:4009(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q55781356(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Paralisia periódica normocalêmica
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata