Doença priônica rara com sintomas neurológicos progressivos, como demência e ataxia. A característica distintiva é a resistência variável das proteínas priônicas anormais à degradação por proteases, o que auxilia no diagnóstico.
Introdução
O que você precisa saber de cara
Visão geral
A Prionopatia sensível a protease, variável (VPSPr) é uma doença priônica rara, caracterizada por uma forma atípica de acúmulo de proteína priônica (PrPSc) que é parcialmente sensível à digestão por protease. Diferentemente de outras doenças priônicas, como a doença de Creutzfeldt-Jakob, a VPSPr apresenta um padrão variável de sensibilidade enzimática, o que torna seu diagnóstico um desafio. A doença afeta o sistema nervoso central e leva a um declínio neurológico progressivo.[1]
Sinais e sintomas
Os sintomas da VPSPr são variáveis e podem incluir alterações cognitivas, distúrbios de movimento, alterações comportamentais e sinais cerebelares. O quadro clínico geralmente evolui de forma mais lenta do que em outras doenças priônicas, mas ainda assim é progressivo e debilitante. A apresentação exata dos sintomas pode diferir entre os pacientes, refletindo a variabilidade da doença.[1]
Causas genéticas
A VPSPr é uma doença priônica esporádica, ou seja, não há uma causa genética hereditária conhecida. Não foram identificados genes específicos associados à condição, e a herança não é estabelecida. A doença está relacionada ao acúmulo anormal da proteína priônica (PrP) no cérebro, mas o mecanismo exato que desencadeia esse processo ainda não é completamente compreendido.[1][3]
Diagnóstico
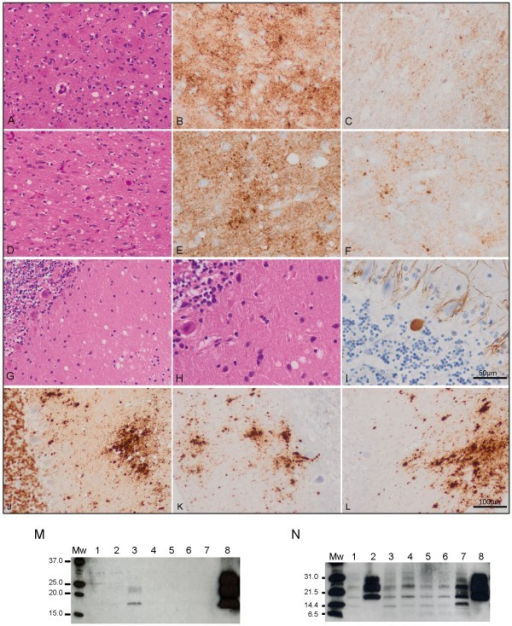
O diagnóstico da VPSPr é complexo e baseia-se na combinação de achados clínicos, exames de imagem (como ressonância magnética), análise do líquido cefalorraquidiano (incluindo a detecção de proteína 14-3-3) e, principalmente, na análise neuropatológica do tecido cerebral, que demonstra o acúmulo de PrPSc com sensibilidade variável à protease. Testes genéticos não são aplicáveis, pois não há gene associado. A classificação CID-10 para a condição é A81.8 (outras doenças priônicas do sistema nervoso central).[1][3]
Tratamento e manejo
Atualmente, não existe tratamento curativo para a VPSPr. O manejo é focado no alívio dos sintomas e no suporte ao paciente e à família, incluindo cuidados paliativos, fisioterapia, terapia ocupacional e suporte nutricional. Não há medicamentos aprovados especificamente para a doença. No Brasil, a condição não possui cobertura específica pelo Sistema Único de Saúde (SUS) para procedimentos ou medicamentos relacionados.[1]
Prognóstico e qualidade de vida
O prognóstico da VPSPr é reservado, com progressão inevitável dos sintomas neurológicos ao longo de meses a anos. A qualidade de vida é significativamente afetada, e o suporte multidisciplinar é essencial para ajudar o paciente e seus cuidadores a lidar com os desafios da doença. A sobrevida pode ser variável, mas geralmente é mais longa do que em outras doenças priônicas clássicas.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Doença priônica rara com sintomas neurológicos progressivos, como demência e ataxia. A característica distintiva é a resistência variável das proteínas priônicas anormais à degradação por proteases, o que auxilia no diagnóstico.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Prionopatia sensível a protease, variável (VPSPr) é uma doença priônica rara, caracterizada por uma forma atípica de acúmulo de proteína priônica (PrPSc) que é parcialmente sensível à digestão por protease. Diferentemente de outras doenças priônicas, como a doença de Creutzfeldt-Jakob, a VPSPr apresenta um padrão variável de sensibilidade enzimática, o que torna seu diagnóstico um desafio. A doença afeta o sistema nervoso central e leva a um declínio neurológico progressivo.[1]
Sinais e sintomas
Os sintomas da VPSPr são variáveis e podem incluir alterações cognitivas, distúrbios de movimento, alterações comportamentais e sinais cerebelares. O quadro clínico geralmente evolui de forma mais lenta do que em outras doenças priônicas, mas ainda assim é progressivo e debilitante. A apresentação exata dos sintomas pode diferir entre os pacientes, refletindo a variabilidade da doença.[1]
Causas genéticas
A VPSPr é uma doença priônica esporádica, ou seja, não há uma causa genética hereditária conhecida. Não foram identificados genes específicos associados à condição, e a herança não é estabelecida. A doença está relacionada ao acúmulo anormal da proteína priônica (PrP) no cérebro, mas o mecanismo exato que desencadeia esse processo ainda não é completamente compreendido.[1][3]
Diagnóstico
O diagnóstico da VPSPr é complexo e baseia-se na combinação de achados clínicos, exames de imagem (como ressonância magnética), análise do líquido cefalorraquidiano (incluindo a detecção de proteína 14-3-3) e, principalmente, na análise neuropatológica do tecido cerebral, que demonstra o acúmulo de PrPSc com sensibilidade variável à protease. Testes genéticos não são aplicáveis, pois não há gene associado. A classificação CID-10 para a condição é A81.8 (outras doenças priônicas do sistema nervoso central).[1][3]
Tratamento e manejo
Atualmente, não existe tratamento curativo para a VPSPr. O manejo é focado no alívio dos sintomas e no suporte ao paciente e à família, incluindo cuidados paliativos, fisioterapia, terapia ocupacional e suporte nutricional. Não há medicamentos aprovados especificamente para a doença. No Brasil, a condição não possui cobertura específica pelo Sistema Único de Saúde (SUS) para procedimentos ou medicamentos relacionados.[1]
Prognóstico e qualidade de vida
O prognóstico da VPSPr é reservado, com progressão inevitável dos sintomas neurológicos ao longo de meses a anos. A qualidade de vida é significativamente afetada, e o suporte multidisciplinar é essencial para ajudar o paciente e seus cuidadores a lidar com os desafios da doença. A sobrevida pode ser variável, mas geralmente é mais longa do que em outras doenças priônicas clássicas.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Prionopatia sensível a protease, variável
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Enhanced Sensitivity of a Modified Quaking-Induced Conversion Diagnostic Test for the Broad Detection of Sporadic and Inherited Prion Diseases: A Retrospective Study.
Quaking-induced conversion (QuIC) tests, which detect prion-seeding activity in cerebrospinal fluid (CSF), have markedly advanced the antemortem diagnosis of prion diseases such as Creutzfeldt-Jakob disease (CJD). These tests provide high diagnostic accuracy and enable timely differentiation from other rapidly progressive neurodegenerative disorders. However, a key limitation of current QuIC tests are the reduced sensitivity in detecting inherited prion diseases and rare sporadic subtypes, including variably protease-sensitive prionopathy (VPSPr). To address this gap, we evaluated a simplified QuIC test, end-point QuIC (EP-QuIC), incorporating a novel recombinant prion protein substrate derived from the North American deer mouse (Peromyscus maniculatus). The diagnostic performance of the modified QuIC test was evaluated using CSF samples from 61 sporadic CJD, 50 inherited prion disease, and 5 VPSPr cases. EP-QuIC with the deer mouse substrate achieved 96.6% sensitivity (111/116) and 100% specificity (35/35), outperforming both standard EP-QuIC (87.1%) and next-generation (IQ-CSF) real-time-QuIC (72.4%) across the same cohort. Notably, this enhanced assay detected inherited mutations, such as D178N, that were previously undetectable with existing diagnostic tests. These findings demonstrate that adapting EP-QuIC with an optimized substrate, termed enhanced sensitivity QuIC (ES-QuIC), significantly improves diagnostic performance for inherited and atypical prion diseases. By expanding the diagnostic reach of QuIC tests, this study strengthens antemortem surveillance, reduces reliance on postmortem confirmation, and improves opportunities for early intervention and clinical trial enrollment, particularly for genetic cases most likely to benefit from emerging therapeutic strategies. ANN NEUROL 2026.
[Prion diseases : Creutzfeldt-Jakob and differential diagnoses].
Prions are unprecedented infectious pathogens that cause a group of rare and inevitably fatal neurodegenerative diseases, affecting approximately 1 person per 1 million inhabitants worldwide each year. These diseases include Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), kuru, fatal insomnia (FI) and variably protease-sensitive prionopathy (VPSPr), all of which involve a conformational change of normal cellular prion protein (PrPC) into abnormal scrapie prion protein (PrPSc) through a posttranslational process. This structural change is associated with profound alterations in the physicochemical properties of PrPC, making the molecule resistant to proteolysis. The conformational alteration of PrPC can occur either due to spontaneous conversion, dominant mutations in the prion protein gene (PRNP) which codes for PrPC or due to an infection with the pathogenic isoform PrPSc from exogenous sources. There is general consensus that PrPC serves as the substrate for the conversion to abnormal PrPSc. The latter multiplies exponentially and accumulates in the brain, forming deposits which are associated with the neurodegenerative changes. Although the understanding of the main causes of prion-induced neurodegeneration is still limited, the spread of PrPSc and neurotoxic signal transfer in the pathogenic process of prions appear to show an interaction. Prion diseases sometimes have long incubation times but also short clinical trajectories. Sporadic and genetic forms occur worldwide of which genetic forms are associated with mutations in PRNP. Zoonotic forms of prion diseases are also associated with bovine diseases. Substantial progress has been made in the diagnosis of these disorders and the diagnostics include magnetic resonance imaging (MRI) and laboratory investigations, particularly of the cerebrospinal fluid. Prionen sind beispiellose infektiöse Krankheitserreger, die eine Gruppe seltener und unweigerlich tödlicher neurodegenerativer Erkrankungen verursachen, die weltweit jährlich etwa 1 Person pro 1 Mio. Einwohner betreffen. Zu diesen Erkrankungen gehören die Creutzfeld-Jacob-Krankheit (CJK), Gerstmann-Sträussler-Scheinker-Syndrom (GSS), Kuru, fatale Schlaflosigkeit (FI) und variable Protease-sensitive Prionopathie (VPSPr), die alle eine konformationelle Veränderung des normalen zellulären Prionproteins (PrPC) in das abnormale Scrapie-Prionprotein (PrPSc) durch einen posttranslationalen Prozess beinhalten. Diese strukturelle Veränderung geht mit tiefgreifenden Veränderungen in den physikochemischen Eigenschaften von PrPC einher, wodurch das Molekül widerstandsfähig gegen Proteolyse wird. Die konformationelle Veränderung von PrPC kann entweder durch spontane Umwandlung, dominante Mutationen im Prionprotein(PRNP)-Gen, das für PrPC codiert, oder durch eine Infektion mit der pathogenen Isoform PrPsc aus exogenen Quellen entstehen. Es besteht allgemeine Einigkeit darüber, dass PrPC als Substrat für die Umwandlung zu abnormalem PrPSc dient. Letzteres vermehrt sich exponentiell und sammelt sich im Gehirn, wodurch Ablagerungen entstehen, die mit den neurodegenerativen Veränderungen verbunden sind. Obwohl das Verständnis der Hauptursachen der prioninduzierten Neurodegeneration noch begrenzt ist, scheint zwischen der Ausbreitung von PrPSc und neurotoxischer Signalübertragung im pathogenen Prozess der Prionen eine Wechselwirkung zu bestehen. Prionenerkrankungen weisen z. T. lange Inkubationszeiten auf und andererseits kurze klinische Verläufe. Weltweit kommen sporadische und genetische Formen vor, von denen genetische Formen mit Mutationen in PRNP assoziiert sind. Zoonotische Formen von Prionenerkrankungen sind u. a. mit Rinderkrankheiten verbunden. Bei der Diagnose dieser Störungen wurden erhebliche Fortschritte erzielt, die Diagnose umfassen Magnetresonanztomographie (MRT) und laborchemische Untersuchungen, insbesondere des Liquor cerebrospinalis.
Variably Protease-Sensitive Prionopathy: Two New Cases With Motor Neuron-Dementia Syndrome.
We describe two patients with variably protease-sensitive prionopathy (VPSPr) who developed progressive upper motor neuron symptoms, insomnia, behavioral and cognitive decline, compatible with primary lateral sclerosis associated with frontotemporal dementia (FTD). Neuropathology revealed a spongiform encephalopathy with frontotemporal and pronounced thalamic involvement, associated with fine synaptic abnormal prion protein conformer (PrPSc) deposits, microplaques, and intraneuronal aggregates. Western blot analysis revealed a characteristic VPSPr proteolytic profile, lacking the diglycosylated band. Both patients were methionine homozygous at PRNP codon 129 and carried no pathogenic mutations. These cases illustrate that VPSPr can present with a prominent motor neuron syndrome and FTD features.
Search for a genetic cause of variably protease-sensitive prionopathy.
Variably protease-sensitive prionopathy (VPSPr) is a rare, atypical subtype of prion disease currently classified as sporadic. We performed exome sequencing and targeted sequencing of PRNP non-coding regions on genomic DNA from autopsy-confirmed VPSPr patients (N = 67) in order to search for a possible genetic cause. Our search identified no potentially causal variants for VPSPr. The common polymorphism PRNP M129V was the largest genetic risk factor for VPSPr, with an odds ratio of 7.0. Other variants in and near PRNP exhibited association to VPSPr risk only in proportion to their linkage disequilibrium with M129V, and upstream expression quantitative trait loci showed no evidence of independent association to VPSPr risk. We cannot rule out the possibility of causal variants hiding in genomic regions or classes of genetic variation that our search did not canvas. Nevertheless, our data support the classification of VPSPr as a sporadic prion disease.
Search for a genetic cause of variably protease-sensitive prionopathy.
Variably protease-sensitive prionopathy (VPSPr) is a rare, atypical subtype of prion disease currently classified as sporadic. We performed exome sequencing and targeted sequencing of PRNP non-coding regions on genomic DNA from autopsy-confirmed VPSPr patients (N=67) in order to search for a possible genetic cause. Our search identified no potentially causal variants for VPSPr. The common polymorphism PRNP M129V was the largest genetic risk factor for VPSPr, with an odds ratio of 7.0. Other variants in and near PRNP exhibited association to VPSPr risk only in proportion to their linkage disequilibrium with M129V, and upstream expression quantitative trait loci showed no evidence of independent association to VPSPr risk. We cannot rule out the possibility of causal variants hiding in genomic regions or classes of genetic variation that our search did not canvas. Nevertheless, our data support the classification of VPSPr as a sporadic prion disease.
Publicações recentes
[Prion diseases : Creutzfeldt-Jakob and differential diagnoses].
Enhanced Sensitivity of a Modified Quaking-Induced Conversion Diagnostic Test for the Broad Detection of Sporadic and Inherited Prion Diseases: A Retrospective Study.
🥉 Relato de casoVariably Protease-Sensitive Prionopathy: Two New Cases With Motor Neuron-Dementia Syndrome.
Variably protease-sensitive prionopathy: mass spectrometry analysis of the pathogenic prion protein provides a new perspective.
Search for a genetic cause of variably protease-sensitive prionopathy.
📚 EuropePMC29 artigos no totalmostrando 31
[Prion diseases : Creutzfeldt-Jakob and differential diagnoses].
Radiologie (Heidelberg, Germany)Enhanced Sensitivity of a Modified Quaking-Induced Conversion Diagnostic Test for the Broad Detection of Sporadic and Inherited Prion Diseases: A Retrospective Study.
Annals of neurologyVariably Protease-Sensitive Prionopathy: Two New Cases With Motor Neuron-Dementia Syndrome.
Annals of clinical and translational neurologyVariably protease-sensitive prionopathy: mass spectrometry analysis of the pathogenic prion protein provides a new perspective.
Acta neuropathologica communicationsSearch for a genetic cause of variably protease-sensitive prionopathy.
PLoS pathogensCharacterization of variably protease-sensitive prionopathy by capillary electrophoresis.
Scientific reportsUpdated global epidemiology atlas of human prion diseases.
Frontiers in public healthVariably protease-sensitive prionopathy with methionine homozygosity at codon 129 in the prion protein gene.
BMJ case reportsHuman prion diseases and the prion protein - what is the current state of knowledge?
Translational neuroscienceAn autopsy case of variably protease-sensitive prionopathy with Met/Met homogeneity at codon 129.
Neuropathology : official journal of the Japanese Society of NeuropathologyDevelopment of an Automated Capillary Immunoassay to Detect Prion Glycotypes in Creutzfeldt-Jakob Disease.
Laboratory investigation; a journal of technical methods and pathologyPhenotypic Heterogeneity of Variably Protease-Sensitive Prionopathy: A Report of Three Cases Carrying Different Genotypes at PRNP Codon 129.
VirusesVariably Protease-sensitive Prionopathy in a Middle-aged Man With Rapidly Progressive Dementia.
Cognitive and behavioral neurology : official journal of the Society for Behavioral and Cognitive NeurologyFurther Characterization of Glycoform-Selective Prions of Variably Protease-Sensitive Prionopathy.
Pathogens (Basel, Switzerland)The utility of bank voles for studying prion disease.
Progress in molecular biology and translational scienceDiagnosis of prion diseases by RT-QuIC results in improved surveillance.
NeurologyVariably protease-sensitive prionopathy: A differential diagnostic consideration for dementia.
Neurology. Clinical practiceUnderstanding Prion Strains: Evidence from Studies of the Disease Forms Affecting Humans.
VirusesVariably protease-sensitive prionopathy mimicking frontotemporal dementia.
Neuropathology : official journal of the Japanese Society of NeuropathologyIn Vitro Seeding Activity of Glycoform-Deficient Prions from Variably Protease-Sensitive Prionopathy and Familial CJD Associated with PrPV180I Mutation.
Molecular neurobiologyRecent advances in the histo-molecular pathology of human prion disease.
Brain pathology (Zurich, Switzerland)Variable Protease-Sensitive Prionopathy Transmission to Bank Voles.
Emerging infectious diseasesVariably protease-sensitive prionopathy presenting within ALS/FTD spectrum.
Annals of clinical and translational neurology[Human prion diseases: current issues].
Zhurnal nevrologii i psikhiatrii imeni S.S. KorsakovaVariably protease-sensitive prionopathy.
Handbook of clinical neurologyMolecular Subtyping of PrPres in Human Sporadic CJD Brain Tissue.
Methods in molecular biology (Clifton, N.J.)Biochemical Characterization of Prions.
Progress in molecular biology and translational scienceNeuropathology of Human Prion Diseases.
Progress in molecular biology and translational scienceHuman prion diseases: surgical lessons learned from iatrogenic prion transmission.
Neurosurgical focusClinical update of Jakob-Creutzfeldt disease.
Current opinion in neurologyA case of variably protease-sensitive prionopathy treated with doxycyclin.
Journal of neurology, neurosurgery, and psychiatryAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Prionopatia sensível a protease, variável.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Prionopatia sensível a protease, variável
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Enhanced Sensitivity of a Modified Quaking-Induced Conversion Diagnostic Test for the Broad Detection of Sporadic and Inherited Prion Diseases: A Retrospective Study.
- [Prion diseases : Creutzfeldt-Jakob and differential diagnoses].
- Variably Protease-Sensitive Prionopathy: Two New Cases With Motor Neuron-Dementia Syndrome.
- Search for a genetic cause of variably protease-sensitive prionopathy.
- Search for a genetic cause of variably protease-sensitive prionopathy.
- Variably protease-sensitive prionopathy: mass spectrometry analysis of the pathogenic prion protein provides a new perspective.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:454742(Orphanet)
- MONDO:0018692(MONDO)
- GARD:21894(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q7915745(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Prionopatia sensível a protease, variável
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata