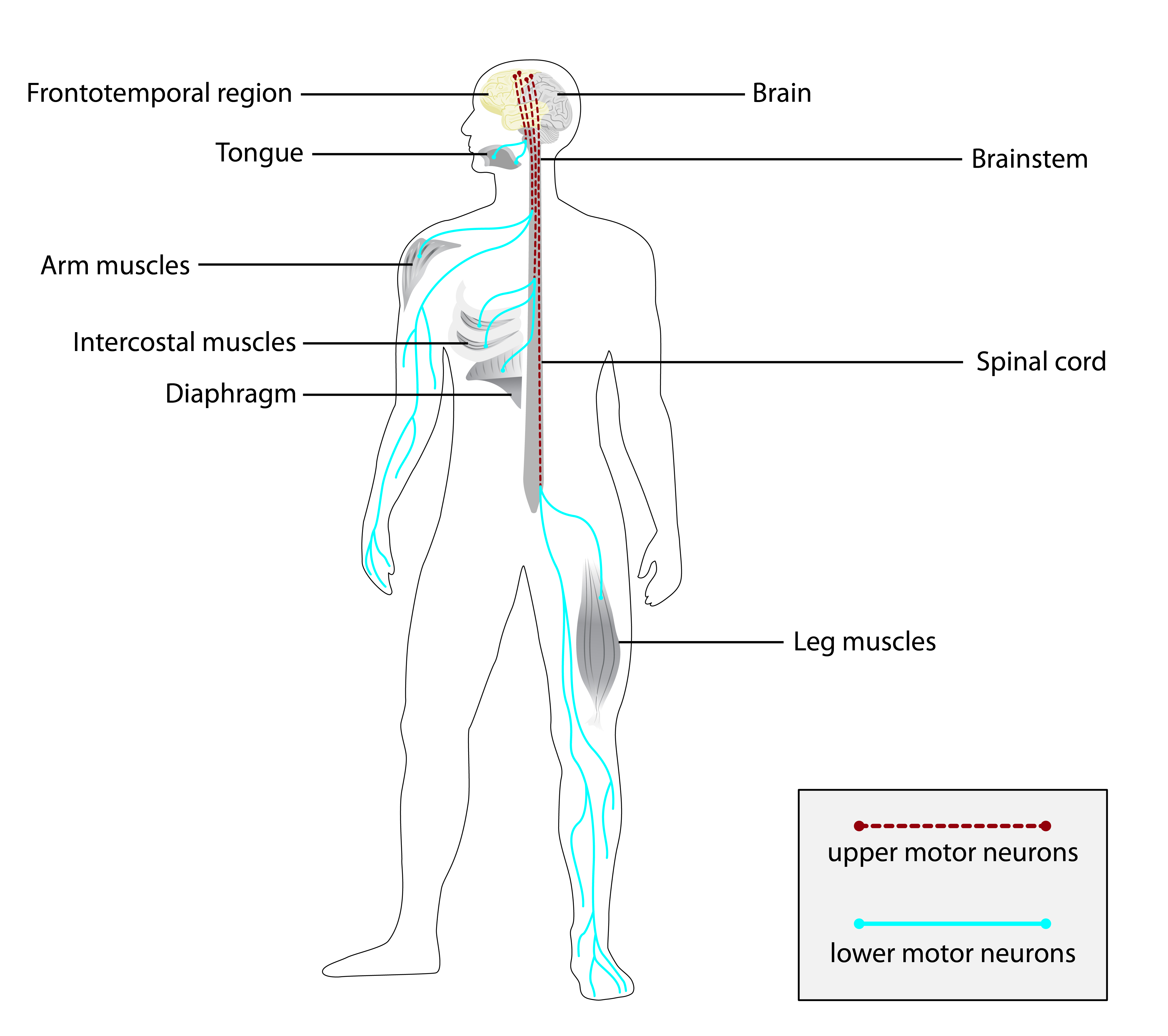
Esclerose lateral amiotrófica (ELA), também conhecida como doença do neurônio motor (DNM) ou doença de Lou Gehrig, é uma doença neurodegenerativa rara e terminal, caracterizada pela perda progressiva de neurônios motores superiores e inferiores que normalmente controlam a contração muscular voluntária. A ELA é a mais comum das doenças do neurônio motor. A ELA frequentemente se manifesta em seus estágios iniciais com rigidez muscular gradual, fasciculações, fraqueza e atrofia. A perda de neurônios motores tipicamente progride até que a capacidade de comer, falar, mover-se e respirar sem suporte mecânico seja perdida. Estima-se que pelo menos 50% das pessoas com ELA experimentem mudanças significativas no pensamento e comportamento, e que 15% dos indivíduos desenvolvam demência frontotemporal.
Introdução
O que você precisa saber de cara
Visão geral
A TARDBP-related predominantly upper-limb distal myopathy é uma doença neuromuscular rara que afeta principalmente os músculos distais dos membros superiores (antebraços, mãos e dedos). A condição é causada por alterações (variantes) no gene TARDBP, que fornece instruções para a produção da proteína TDP-43. Esta proteína desempenha um papel importante na regulação da expressão de outros genes e na manutenção da saúde dos neurônios e das células musculares. A doença foi recentemente reconhecida e classificada na ontologia de doenças raras (MONDO:0979359).[1][2]
Sinais e sintomas
Os principais sintomas envolvem fraqueza muscular e atrofia (perda de massa muscular) que começa nos membros superiores, especialmente nas mãos e antebraços. A fraqueza é distal, ou seja, afeta mais as extremidades do que os músculos próximos ao tronco. Com a progressão da doença, pode haver dificuldade para realizar movimentos finos com as mãos, como escrever, abotoar roupas ou segurar objetos. A fraqueza pode se estender para os membros inferiores em estágios mais avançados, mas o padrão predominante é o comprometimento dos braços e mãos.[1]
Causas genéticas
A doença é causada por variantes patogênicas (mutações) no gene TARDBP (TAR DNA-binding protein 43). Este gene está localizado no cromossomo 1 e codifica a proteína TDP-43, que é essencial para o processamento do RNA em diversos tecidos, incluindo o músculo esquelético. As variantes no TARDBP podem levar ao acúmulo anormal da proteína TDP-43 nas células musculares, contribuindo para a degeneração muscular. Até o momento, 270 variantes genéticas associadas a esta condição foram catalogadas no ClinVar, um banco de dados público de variantes genéticas.[1][3]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sintomas, exames de imagem (como ressonância magnética muscular) e, principalmente, no teste genético molecular. O teste genético identifica variantes patogênicas no gene TARDBP. A análise pode ser feita por sequenciamento de nova geração (painel de genes) ou sequenciamento completo do exoma. O ClinVar registra atualmente 270 variantes associadas a esta condição, o que auxilia na interpretação dos resultados genéticos. Não há um código CID-10 específico para esta doença, e ela não está incluída na tabela de procedimentos do SUS.[1][3]
Tratamento e manejo
Atualmente, não existe cura para a TARDBP-related predominantly upper-limb distal myopathy. O manejo é focado no alívio dos sintomas e na manutenção da função muscular. As abordagens incluem fisioterapia para fortalecer os músculos preservados e melhorar a amplitude de movimento, terapia ocupacional para adaptar as atividades diárias e, em alguns casos, o uso de órteses para auxiliar na função das mãos. Não há medicamentos específicos aprovados para esta condição. O acompanhamento multidisciplinar com neurologista, fisiatra e geneticista é recomendado para monitorar a progressão da doença e oferecer suporte ao paciente e à família.[1]
Prognóstico e qualidade de vida
O prognóstico varia de acordo com a gravidade das variantes genéticas e a idade de início dos sintomas. A doença tende a ser progressiva, mas a velocidade de progressão pode ser lenta em muitos casos. A qualidade de vida pode ser impactada pela perda progressiva da função manual, mas com suporte adequado (fisioterapia, adaptações ergonômicas e acompanhamento psicológico), muitos pacientes conseguem manter independência nas atividades básicas por vários anos. Não há dados disponíveis sobre a prevalência exata ou a expectativa de vida específica para esta condição.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Esclerose lateral amiotrófica (ELA), também conhecida como doença do neurônio motor (DNM) ou doença de Lou Gehrig, é uma doença neurodegenerativa rara e terminal, caracterizada pela perda progressiva de neurônios motores superiores e inferiores que normalmente controlam a contração muscular voluntária. A ELA é a mais comum das doenças do neurônio motor. A ELA frequentemente se manifesta em seus estágios iniciais com rigidez muscular gradual, fasciculações, fraqueza e atrofia. A perda de neurônios motores tipicamente progride até que a capacidade de comer, falar, mover-se e respirar sem suporte mecânico seja perdida. Estima-se que pelo menos 50% das pessoas com ELA experimentem mudanças significativas no pensamento e comportamento, e que 15% dos indivíduos desenvolvam demência frontotemporal.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A TARDBP-related predominantly upper-limb distal myopathy é uma doença neuromuscular rara que afeta principalmente os músculos distais dos membros superiores (antebraços, mãos e dedos). A condição é causada por alterações (variantes) no gene TARDBP, que fornece instruções para a produção da proteína TDP-43. Esta proteína desempenha um papel importante na regulação da expressão de outros genes e na manutenção da saúde dos neurônios e das células musculares. A doença foi recentemente reconhecida e classificada na ontologia de doenças raras (MONDO:0979359).[1][2]
Sinais e sintomas
Os principais sintomas envolvem fraqueza muscular e atrofia (perda de massa muscular) que começa nos membros superiores, especialmente nas mãos e antebraços. A fraqueza é distal, ou seja, afeta mais as extremidades do que os músculos próximos ao tronco. Com a progressão da doença, pode haver dificuldade para realizar movimentos finos com as mãos, como escrever, abotoar roupas ou segurar objetos. A fraqueza pode se estender para os membros inferiores em estágios mais avançados, mas o padrão predominante é o comprometimento dos braços e mãos.[1]
Causas genéticas
A doença é causada por variantes patogênicas (mutações) no gene TARDBP (TAR DNA-binding protein 43). Este gene está localizado no cromossomo 1 e codifica a proteína TDP-43, que é essencial para o processamento do RNA em diversos tecidos, incluindo o músculo esquelético. As variantes no TARDBP podem levar ao acúmulo anormal da proteína TDP-43 nas células musculares, contribuindo para a degeneração muscular. Até o momento, 270 variantes genéticas associadas a esta condição foram catalogadas no ClinVar, um banco de dados público de variantes genéticas.[1][3]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sintomas, exames de imagem (como ressonância magnética muscular) e, principalmente, no teste genético molecular. O teste genético identifica variantes patogênicas no gene TARDBP. A análise pode ser feita por sequenciamento de nova geração (painel de genes) ou sequenciamento completo do exoma. O ClinVar registra atualmente 270 variantes associadas a esta condição, o que auxilia na interpretação dos resultados genéticos. Não há um código CID-10 específico para esta doença, e ela não está incluída na tabela de procedimentos do SUS.[1][3]
Tratamento e manejo
Atualmente, não existe cura para a TARDBP-related predominantly upper-limb distal myopathy. O manejo é focado no alívio dos sintomas e na manutenção da função muscular. As abordagens incluem fisioterapia para fortalecer os músculos preservados e melhorar a amplitude de movimento, terapia ocupacional para adaptar as atividades diárias e, em alguns casos, o uso de órteses para auxiliar na função das mãos. Não há medicamentos específicos aprovados para esta condição. O acompanhamento multidisciplinar com neurologista, fisiatra e geneticista é recomendado para monitorar a progressão da doença e oferecer suporte ao paciente e à família.[1]
Prognóstico e qualidade de vida
O prognóstico varia de acordo com a gravidade das variantes genéticas e a idade de início dos sintomas. A doença tende a ser progressiva, mas a velocidade de progressão pode ser lenta em muitos casos. A qualidade de vida pode ser impactada pela perda progressiva da função manual, mas com suporte adequado (fisioterapia, adaptações ergonômicas e acompanhamento psicológico), muitos pacientes conseguem manter independência nas atividades básicas por vários anos. Não há dados disponíveis sobre a prevalência exata ou a expectativa de vida específica para esta condição.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição.
RNA-binding protein that is involved in various steps of RNA biogenesis and processing (PubMed:23519609). Preferentially binds, via its two RNA recognition motifs RRM1 and RRM2, to GU-repeats on RNA molecules predominantly localized within long introns and in the 3'UTR of mRNAs (PubMed:23519609, PubMed:24240615, PubMed:24464995). In turn, regulates the splicing of many non-coding and protein-coding RNAs including proteins involved in neuronal survival, as well as mRNAs that encode proteins relev
NucleusCytoplasmCytoplasm, Stress granuleMitochondrion
Amyotrophic lateral sclerosis 10
A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases.
Variantes genéticas (ClinVar)
270 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — TARDBP-related predominantly upper-limb distal myopathy
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para TARDBP-related predominantly upper-limb distal myopathy.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para TARDBP-related predominantly upper-limb distal myopathy
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:700154(Orphanet)
- MONDO:0979359(MONDO)
- Variantes catalogadas(ClinVar)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
TARDBP-related predominantly upper-limb distal myopathy
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO