Epilepsia mioclônica em encefalopatias não-progressivas é um distúrbio neurológico caracterizado por espasmos musculares súbitos e generalizados (mioclonias) associados a epilepsia, em um quadro de dano cerebral não evolutivo. Geralmente se manifesta na infância, com desenvolvimento cognitivo e motor prejudicados, mas sem piora progressiva das lesões cerebrais.
Introdução
O que você precisa saber de cara
Visão geral
A epilepsia mioclônica em encefalopatias não-progressivas é uma síndrome epiléptica rara que se manifesta em bebês e crianças pequenas. A principal característica são crises mioclônicas recorrentes e prolongadas (estado de mal mioclônico), que ocorrem em crianças com uma encefalopatia que não piora com o tempo. Essas crises vêm acompanhadas de alterações transitórias e recorrentes nas funções motoras, cognitivas e/ou comportamentais.[1]
Sinais e sintomas
Os sintomas começam na primeira infância (infância ou início da idade escolar). As crianças apresentam episódios prolongados de abalos musculares involuntários (mioclonias) que podem durar horas ou dias. Durante esses episódios, podem ocorrer dificuldades motoras transitórias, confusão mental ou alterações no comportamento. Entre as crises, a criança geralmente retorna ao seu estado habitual, pois a encefalopatia de base não é progressiva.[1]
Causas genéticas
Diagnóstico
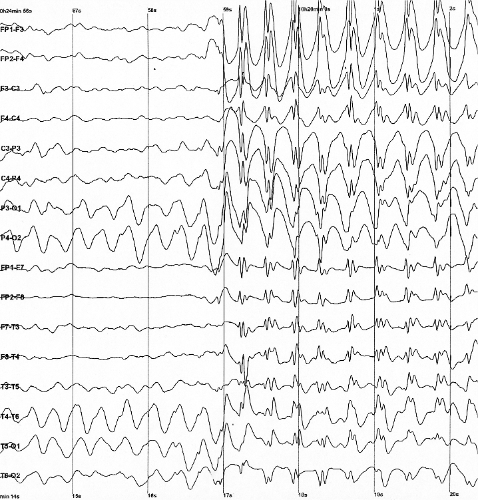
O diagnóstico é clínico, baseado na observação das crises mioclônicas prolongadas em uma criança com encefalopatia não-progressiva. Exames como o eletroencefalograma (EEG) podem auxiliar na confirmação. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura parcial para o diagnóstico, incluindo o sequenciamento completo do exoma (WES) e atendimento em reabilitação para doenças raras.[1]
Tratamento e manejo
O manejo é focado no controle das crises mioclônicas e no suporte às alterações transitórias. Medicamentos antiepilépticos podem ser utilizados, mas a escolha deve ser individualizada por um médico especialista. No Brasil, o SUS oferece cobertura parcial para o tratamento, incluindo o acesso a medicamentos padronizados e atendimento em reabilitação. É fundamental que o tratamento seja acompanhado por uma equipe multidisciplinar (neurologista, pediatra, fisioterapeuta, terapeuta ocupacional).[1]
Tratamentos citados na literatura
Não há dados disponíveis na literatura científica (fonte PubTator3) sobre tratamentos específicos para esta condição. As informações sobre medicamentos listados em bases de dados gerais (como ácido aminohidroxibutírico, diclofenamida, etossuximida, felbamato, fosfenitoína, lamotrigina, metarbital, fenacemida, piridoxal, stiripentol, topiramato, ácido valproico, valpromida e zonisamida) são associações mineradas da literatura científica, mas não representam recomendações de tratamento para esta síndrome em particular. Consulte sempre um médico neurologista.[1]
Prognóstico e qualidade de vida
Como a encefalopatia de base não é progressiva, o prognóstico geral é de estabilidade ao longo do tempo. As crises mioclônicas podem ser controladas com tratamento adequado, e as alterações motoras, cognitivas e comportamentais tendem a ser transitórias. O acompanhamento regular com neurologista e equipe multidisciplinar é essencial para manter a qualidade de vida da criança e da família.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Epilepsia mioclônica em encefalopatias não-progressivas é um distúrbio neurológico caracterizado por espasmos musculares súbitos e generalizados (mioclonias) associados a epilepsia, em um quadro de dano cerebral não evolutivo. Geralmente se manifesta na infância, com desenvolvimento cognitivo e motor prejudicados, mas sem piora progressiva das lesões cerebrais.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A epilepsia mioclônica em encefalopatias não-progressivas é uma síndrome epiléptica rara que se manifesta em bebês e crianças pequenas. A principal característica são crises mioclônicas recorrentes e prolongadas (estado de mal mioclônico), que ocorrem em crianças com uma encefalopatia que não piora com o tempo. Essas crises vêm acompanhadas de alterações transitórias e recorrentes nas funções motoras, cognitivas e/ou comportamentais.[1]
Sinais e sintomas
Os sintomas começam na primeira infância (infância ou início da idade escolar). As crianças apresentam episódios prolongados de abalos musculares involuntários (mioclonias) que podem durar horas ou dias. Durante esses episódios, podem ocorrer dificuldades motoras transitórias, confusão mental ou alterações no comportamento. Entre as crises, a criança geralmente retorna ao seu estado habitual, pois a encefalopatia de base não é progressiva.[1]
Causas genéticas
Diagnóstico
O diagnóstico é clínico, baseado na observação das crises mioclônicas prolongadas em uma criança com encefalopatia não-progressiva. Exames como o eletroencefalograma (EEG) podem auxiliar na confirmação. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura parcial para o diagnóstico, incluindo o sequenciamento completo do exoma (WES) e atendimento em reabilitação para doenças raras.[1]
Tratamento e manejo
O manejo é focado no controle das crises mioclônicas e no suporte às alterações transitórias. Medicamentos antiepilépticos podem ser utilizados, mas a escolha deve ser individualizada por um médico especialista. No Brasil, o SUS oferece cobertura parcial para o tratamento, incluindo o acesso a medicamentos padronizados e atendimento em reabilitação. É fundamental que o tratamento seja acompanhado por uma equipe multidisciplinar (neurologista, pediatra, fisioterapeuta, terapeuta ocupacional).[1]
Tratamentos citados na literatura
Não há dados disponíveis na literatura científica (fonte PubTator3) sobre tratamentos específicos para esta condição. As informações sobre medicamentos listados em bases de dados gerais (como ácido aminohidroxibutírico, diclofenamida, etossuximida, felbamato, fosfenitoína, lamotrigina, metarbital, fenacemida, piridoxal, stiripentol, topiramato, ácido valproico, valpromida e zonisamida) são associações mineradas da literatura científica, mas não representam recomendações de tratamento para esta síndrome em particular. Consulte sempre um médico neurologista.[1]
Prognóstico e qualidade de vida
Como a encefalopatia de base não é progressiva, o prognóstico geral é de estabilidade ao longo do tempo. As crises mioclônicas podem ser controladas com tratamento adequado, e as alterações motoras, cognitivas e comportamentais tendem a ser transitórias. O acompanhamento regular com neurologista e equipe multidisciplinar é essencial para manter a qualidade de vida da criança e da família.[1]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Epilepsia mioclônica em encefalopatias não-progressivas
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Familial adult myoclonus epilepsy: Clinical findings, disease course, and comorbidities.
Familial adult myoclonus epilepsy (FAME) is an autosomal dominant condition characterized by the association of myoclonic tremor and epilepsy mainly with onset in adulthood. The clinical course is non-progressive or slowly progressive, as epilepsy is commonly controlled with appropriate antiseizure medication and individuals have a normal life expectancy. However, the myoclonus severity increases with age and leads to some degree of disability in the elderly. Because the non-coding repeat expansions responsible for FAME are not detected by routine genetic tests being used at this time, a clinical diagnosis accompanied by neurophysiological testing remains essential to guide the geneticist on the selection of the specific genetic technique.
Myoclonic status epilepticus in non-progressive encephalopathies within the GRIN2A-associated epilepsy-aphasia spectrum.
Monoamine neurotransmitters in early epileptic encephalopathies: New insights into pathophysiology and therapy.
To study neurotransmitter status in children with early epileptic and developmental and epileptic encephalopathy (DEE) and to explore the clinical response to dopaminergic and serotoninergic therapies in a group of patients. Two hundred and five patients (111 males [54.1.%] and 94 females [45.9%], mean age 10 months at the onset of epilepsy [SD 1 year 1 month], range 0-3 year) with epileptic encephalopathy/DEE were recruited, including those with West syndrome, Ohtahara syndrome, early myoclonic encephalopathy, epilepsy of infancy with migrating focal seizures, myoclonic encephalopathy in non-progressive disorders, infantile spasms, Doose syndrome, Lennox-Gastaut syndrome, Landau-Kleffner syndrome, and those unclassified. Cerebrospinal fluid (CSF) neurotransmitter studies and patients' medical records were reviewed. Additionally, we present clinical data of 10 patients with low CSF neurotransmitter levels who received dopaminergic/serotoninergic treatments. Abnormal neurotransmitter values were identified in 68 (33%) patients. 5-Hydroxyindoleacetic acid (5-HIAA) deficit was the most prevalent alteration (91%). Low CSF 5-HIAA levels were significantly higher in 1- to 3-year-old children. A negative significant correlation was found between 5-HIAA levels and epilepsy duration before CSF study (Spearman's ρ=-0.191, p=0.007). Abnormalities in deep grey matter were associated with low levels of CSF homovanillic acid and 5-HIAA. Ten patients with low CSF neurotransmitter levels received dopamine and/or serotonin therapies. Six of them showed initial decrease of seizure frequency and severity and maintained improvement in some neurodevelopmental skills. A considerable number of patients showed neurotransmitter abnormalities. Age at seizure onset and duration of epilepsy before CSF study were the principal factors related to neurotransmitter depletion. Early monoamine supplementation would seem advisable as a neuroprotective strategy. 5-Hydroxyindoleacetic acid homeostasis is especially vulnerable in patients with epileptic encephalopathy/developmental and epileptic encephalopathy. Age of seizure onset and duration of epilepsy are determinants of neurotransmitter depletion.
Clinical and genetic study of developmental and epileptic encephalopathy in Argentinean pediatric patients.
The aim of this study was to extend our knowledge of the genetic background of Argentinean pediatric patients with developmental and epileptic encephalopathy (DEE) applying a next generation sequencing (NGS) panel. Thirty one patients with DEE were studied, including these phenotypes: Dravet syndrome (n:7), Dravet like syndrome (n:3), West syndrome (WS) (n:6), WS that evolved to Lennox-Gastaut syndrome (LGS) (n:4), epilepsy of infancy with migrating focal seizures (n:2), continuous spikes and waves during slow sleep evolving to LGS (n:1), LGS (n:1), myoclonic status in non-progressive encephalopathy (n:1), myoclonic atonic epilepsy (n:1), epileptic encephalopathy with multifocal spikes (n:1) and unclassified epileptic encephalopathy (n:4). Fifty-two genes frequently associated with DEE were studied by NGS in genomic DNA from peripheral blood. Relevant variants were detected in 12 cases; 6 novel pathogenic or likely pathogenic variants, 6 previously reported as pathogenic and 1 variant of unknown significance. Single-nucleotide heterozygous variants were identified in the SCN1A (5), GABRG2 (1), STXBP1 (2) genes, a mosaic variant in SCN2A (1) and a homozygous variant in SCN1B (1). Additionally, a heterozygous deletion involving the SCN1A, SCN2A and SCN3A genes (1), and the most frequent triplet repeat expansion in the ARX gene (1) were detected. Genetic diagnosis was made in 39% of patients. We emphasize the importance of considering mosaic variants, copy number variants and hereditary forms when designing and interpreting molecular studies, to optimize diagnosis and management of patients. Approximately 42% of the detected variants were novel, expanding the knowledge of the molecular basis of DEEs in Latin-American patients. Introducción: El objetivo del estudio fue ampliar el conocimiento de las bases moleculares de las encefalopatías epilépticas y del desarrollo (EED) en pacientes pediátricos argentinos aplicando un panel de secuenciación de nueva generación (NGS). Métodos: Se analizaron 31 pacientes con los fenotipos clínicos de síndrome de Dravet (n:7), síndrome símil Dravet (n:3), síndrome de West (SW) (n:6), SW que evoluciona a síndrome de Lennox Gastaut (SLG)(N:4), epilepsia de la infancia con crisis focales migratorias (n:2), actividad de punta onda continua durante el sueño que evolucionan a SLG (n:1), SLG (n:1), encefalopatía no progresiva con estatus mioclónico (n:1), epilepsia mioclónica atónica (n:1), encefalopatía epiléptica con espigas multifocales (n:1) y encefalopatía epiléptica indeterminada (n:4). Se estudiaron los 52 genes más frecuentemente asociados a EED a través de NGS, en ADN extraído de sangre periférica. Resultados: Se identificaron variantes relevantes en 12 casos, de las cuales 5 fueron nuevas y 6 previamente reportadas como patogénicas o posiblemente patogénicas, mientras que una variante fue clasificada como de significado incierto. Variantes heterocigotas, de nucleótido único, se identificaron en los genes SCN1A (5), GABRG2 (1), STXBP1 (2), una variante en mosaico en SCN2A (1) y otra homocigota en SCN1B (1). Además, se detectó una deleción que involucra a los genes SCN1A, SCN2A y SCN3A (1) y la expansión de repeticiones de tripletes más frecuente en el gen ARX (1). Discusión: Se alcanzó el diagnóstico molecular en el 39% de los pacientes. Remarcamos la importancia de considerar variantes en mosaico, variantes en el número de copias y formas heredadas al momento de diseñar e interpretar los estudios moleculares, de tal forma de optimizar el diagnóstico y seguimiento de los pacientes con EED. Cabe destacar, que el 42% de las variantes detectadas fueron nuevas, ampliando nuestro conocimiento sobre las bases moleculares de las EED en población latino americana.
Fifteen-year follow-up of a patient with a DHDDS variant with non-progressive early onset myoclonic tremor and rare generalized epilepsy.
Generalized epilepsy and tremor phenotypes have been reported in some genetic disorders. Among them benign adult familial myoclonus epilepsy (BAFME) has been confirmed as a clearly defined clinical and genetic entity. On the other hand, non-progressive tremor and generalized epilepsy phenotypes have also been reported in patients with DHDDS variants. We report on a long term follow-up of patient with de novo missense variant of DHDDS, who revealed non progressive nature. This 18-year-old woman presented non-progressive tremor since her early infancy. She had rare seizures. Her tremor was considered as cortical myoclonic tremor with giant somatosensory evoked potentials. In patients with early onset, non-progressive tremor and rare generalized epilepsy phenotypes, DHDDS variants may be considered in the genetic differential diagnosis.
Publicações recentes
Myoclonic status epilepticus in non-progressive encephalopathies within the GRIN2A-associated epilepsy-aphasia spectrum.
Familial adult myoclonus epilepsy: Clinical findings, disease course, and comorbidities.
Clinical and genetic study of developmental and epileptic encephalopathy in Argentinean pediatric patients.
Monoamine neurotransmitters in early epileptic encephalopathies: New insights into pathophysiology and therapy.
Fifteen-year follow-up of a patient with a DHDDS variant with non-progressive early onset myoclonic tremor and rare generalized epilepsy.
📚 EuropePMCmostrando 12
Myoclonic status epilepticus in non-progressive encephalopathies within the GRIN2A-associated epilepsy-aphasia spectrum.
SeizureFamilial adult myoclonus epilepsy: Clinical findings, disease course, and comorbidities.
EpilepsiaClinical and genetic study of developmental and epileptic encephalopathy in Argentinean pediatric patients.
MedicinaMonoamine neurotransmitters in early epileptic encephalopathies: New insights into pathophysiology and therapy.
Developmental medicine and child neurologyFifteen-year follow-up of a patient with a DHDDS variant with non-progressive early onset myoclonic tremor and rare generalized epilepsy.
Brain & development[Genetically determined epileptic encephalopathies].
MedicinaBerardinelli-Seip syndrome and progressive myoclonus epilepsy.
Epileptic disorders : international epilepsy journal with videotapeBenign and severe early-life seizures: a round in the first year of life.
Italian journal of pediatricsThe ketogenic diet in patients with myoclonic status in non-progressive encephalopathy.
SeizureGray Matter Loss and Related Functional Connectivity Alterations in A Chinese Family With Benign Adult Familial Myoclonic Epilepsy.
MedicineAudiogenic reflex seizures in cats.
Journal of feline medicine and surgery[Recent advance in research of benign adult familial myoclonus epilepsy (BAFME): is BAFME a progressive disorder?].
Rinsho shinkeigaku = Clinical neurologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Epilepsia mioclônica em encefalopatias não-progressivas.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Epilepsia mioclônica em encefalopatias não-progressivas
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Ainda não achamos doenças com sintomas parecidos o suficiente.
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Familial adult myoclonus epilepsy: Clinical findings, disease course, and comorbidities.
- Myoclonic status epilepticus in non-progressive encephalopathies within the GRIN2A-associated epilepsy-aphasia spectrum.
- Monoamine neurotransmitters in early epileptic encephalopathies: New insights into pathophysiology and therapy.
- Clinical and genetic study of developmental and epileptic encephalopathy in Argentinean pediatric patients.
- Fifteen-year follow-up of a patient with a DHDDS variant with non-progressive early onset myoclonic tremor and rare generalized epilepsy.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:86913(Orphanet)
- MONDO:0019488(MONDO)
- Epilepsia(PCDT · Ministério da Saúde)
- GARD:19088(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q55788681(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Epilepsia mioclônica em encefalopatias não-progressivas
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos aprovados FDA
- fonte: FDA OpenFDA
- Reposicionamento
- fonte: Drug Repurposing Hub