Uma displasia esquelética rara e letal, caracterizada por nanismo grave com membros curtos, articulações deslocadas, pés tortos congênitos, traços faciais distintos e achados de raio-X que mostram ossos longos dos braços e pernas com ossificação insuficiente e desenvolvimento anormal, apresentando uma curvatura que lembra um bumerangue.
Introdução
O que você precisa saber de cara
Uma displasia esquelética rara e letal, caracterizada por nanismo grave com membros curtos, articulações deslocadas, pés tortos congênitos, traços faciais distintos e achados de raio-X que mostram ossos longos dos braços e pernas com ossificação insuficiente e desenvolvimento anormal, apresentando uma curvatura que lembra um bumerangue.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 17 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 30 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Not applicable.
Curadoria gene-doença
fontes oficiaisConnects cell membrane constituents to the actin cytoskeleton. May promote orthogonal branching of actin filaments and links actin filaments to membrane glycoproteins. Anchors various transmembrane proteins to the actin cytoskeleton. Interaction with FLNA may allow neuroblast migration from the ventricular zone into the cortical plate. Various interactions and localizations of isoforms affect myotube morphology and myogenesis. Isoform 6 accelerates muscle differentiation in vitro
Cytoplasm, cell cortexCytoplasm, cytoskeletonCytoplasm, cytoskeleton, stress fiberCytoplasm, myofibril, sarcomere, Z line
Variantes genéticas (ClinVar)
508 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 45 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Displasia boomerang
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Cell-Dependent Pathogenic Roles of Filamin B in Different Skeletal Malformations.
Mutations of filamin B (FLNB) gene can lead to a spectrum of autosomal skeletal malformations including spondylocarpotarsal syndrome (SCT), Larsen syndrome (LRS), type I atelosteogenesis (AO1), type III atelosteogenesis (AO3), and boomerang dysplasia (BD). Among them, LRS is milder while BD causes a more severe phenotype. However, the molecular mechanism underlying the differences in clinical phenotypes of different FLNB variants has not been fully determined. Here, we presented two patients suffering from autosomal dominant LRS and autosomal recessive vitamin D-dependent rickets type IA (VDDR-IA). Whole-exome sequencing revealed two novel missense variants in FLNB, c.4846A>G (p.T1616A) and c.7022T>G (p.I2341R), which are located in repeat 15 and 22 of filamin B, respectively. The expression of FLNBI2341R in the muscle tissue from our LRS patient was remarkably increased. And in vitro studies showed that both variants led to a lack of filopodia and accumulation of the mutants in the perinuclear region in HEK293 cells. We also found that c.4846A>G (p.T1616A) and c.7022T>G (p.I2341R) regulated endochondral osteogenesis in different ways. c.4846A>G (p.T1616A) activated AKT pathways through inhibiting SHIP2, suppressed the Smad3 pathway, and impaired the expression of Runx2 in both Saos-2 and ATDC5 cells. c.7022T>G (p.I2341R) activated both AKT and Smad3 pathways and increased the expression of Runx2 in Saos-2 cells, while in ATDC5 cells it activated AKT pathways through inhibiting SHIP2, suppressed the Smad3 pathway, and reduced the expression of Runx2. Our study demonstrated the pathogenic mechanisms of two novel FLNB variants in two different clinical settings and proved that FLNB variants could not only directly cause skeletal malformations but also worsen skeletal symptoms in the setting of other skeletal diseases. Besides, FLNB variants differentially affect skeletal development which contributes to clinical heterogeneity of FLNB-related disorders.
Deciphering the Role of Filamin B Calponin-Homology Domain in Causing the Larsen Syndrome, Boomerang Dysplasia, and Atelosteogenesis Type I Spectrum Disorders via a Computational Approach.
Filamins (FLN) are a family of actin-binding proteins involved in regulating the cytoskeleton and signaling phenomenon by developing a network with F-actin and FLN-binding partners. The FLN family comprises three conserved isoforms in mammals: FLNA, FLNB, and FLNC. FLNB is a multidomain monomer protein with domains containing an actin-binding N-terminal domain (ABD 1-242), encompassing two calponin-homology domains (assigned CH1 and CH2). Primary variants in FLNB mostly occur in the domain (CH2) and surrounding the hinge-1 region. The four autosomal dominant disorders that are associated with FLNB variants are Larsen syndrome, atelosteogenesis type I (AOI), atelosteogenesis type III (AOIII), and boomerang dysplasia (BD). Despite the intense clustering of FLNB variants contributing to the LS-AO-BD disorders, the genotype-phenotype correlation is still enigmatic. In silico prediction tools and molecular dynamics simulation (MDS) approaches have offered the potential for variant classification and pathogenicity predictions. We retrieved 285 FLNB missense variants from the UniProt, ClinVar, and HGMD databases in the current study. Of these, five and 39 variants were located in the CH1 and CH2 domains, respectively. These variants were subjected to various pathogenicity and stability prediction tools, evolutionary and conservation analyses, and biophysical and physicochemical properties analyses. Molecular dynamics simulation (MDS) was performed on the three candidate variants in the CH2 domain (W148R, F161C, and L171R) that were predicted to be the most pathogenic. The MDS analysis results showed that these three variants are highly compact compared to the native protein, suggesting that they could affect the protein on the structural and functional levels. The computational approach demonstrates the differences between the FLNB mutants and the wild type in a structural and functional context. Our findings expand our knowledge on the genotype-phenotype correlation in FLNB-related LS-AO-BD disorders on the molecular level, which may pave the way for optimizing drug therapy by integrating precision medicine.
Fetal magnetic resonance imaging of skeletal dysplasias.
Fetal magnetic resonance imaging (MRI) is obtained for prenatal diagnosis and prognostication of skeletal dysplasias; however, related literature is limited. The purpose of this study was to define the utility of fetal MRI for skeletal dysplasias and to report MRI findings associated with specific diagnoses. This retrospective study was approved by the institutional review board; informed consent was waived. Women referred for suspected fetal skeletal dysplasia who underwent MRI between January 2003 and December 2018 were included. Definitive diagnoses were determined by genetic testing, autopsy, physical examination and/or postnatal/postmortem imaging. Fetal MRI examinations and reports were reviewed. Descriptive statistics were used to summarize imaging findings. Eighty-nine women were referred for fetal MRI for possible skeletal dysplasia. Forty-three (48%) were determined to have a diagnosis other than skeletal dysplasia and nine were excluded for lack of specific skeletal dysplasia diagnosis. Thirty-seven cases of skeletal dysplasia with available fetal MRI and specific diagnosis were included for analysis. Diagnoses included achondrogenesis (n=2), achondroplasia (n=5), Boomerang dysplasia (n=1), campomelic dysplasia (n=2), Jeune syndrome (n=1), Kniest dysplasia (n=1), osteogenesis imperfecta (n=15) and thanatophoric dysplasia (n=10). A specific skeletal dysplasia diagnosis was mentioned in 17/37 (46%) of MRI imaging reports and correct for 14/17 (82%). MRI findings were reported for each specific skeletal dysplasia diagnosis. Fetal MRI is a useful diagnostic tool for skeletal dyplasias and excluded the diagnosis in nearly half of referred pregnancies. In addition to providing fetal lung volumes, fetal MRI demonstrates findings of the brain in achondroplasia and thanatophoric dysplasia, of the spine in achondroplasia and achondrogenesis, of the calvarium in osteogenesis imperfecta and thanatophoric dysplasia, and of the cartilage in Kniest dysplasia.
Seven additional families with spondylocarpotarsal synostosis syndrome with novel biallelic deleterious variants in FLNB.
The location and/or type of variants in FLNB result in a spectrum of osteochondrodysplasias ranging from mild forms, like spondylocarpotarsal synostosis syndrome and Larsen syndrome, to severe perinatal lethal forms, such as atelosteogenesis I and III and Boomerang dysplasia. Spondylocarpotarsal synostosis syndrome is characterized by disproportionate short stature, vertebral anomalies and fusion of carpal and tarsal bones. Biallelic loss-of-function variants in FLNB are known to cause spondylocarpotarsal synostosis syndrome and 9 families and 9 pathogenic variants have been reported so far. We report clinical features of 10 additional patients from 7 families with spondylocarpotarsal synostosis syndrome due to 7 novel deleterious variants in FLNB, thus expanding the clinical and molecular repertoire of spondylocarpotarsal synostosis syndrome. Our report validates key clinical (fused thoracic vertebrae and carpal and tarsal coalition) and molecular (truncating variants in FLNB) characteristics of this condition.
Comparative analysis of the two extremes of FLNB-mutated autosomal dominant disease spectrum: from clinical phenotypes to cellular and molecular findings.
Non-randomly distributed missense mutations of Filamin B (FLNB) can lead to a spectrum of autosomal dominant-inherited skeletal malformations caused by bone hypoplasia, including Larsen syndrome (LS), atelosteogenesi-I (AO-I), atelosteogenesi-I (AO-III) and boomerang dysplasia (BD). Among this spectrum of diseases, LS causes a milder hypoplasia of the skeletal system, compared to BD's much more severe symptoms. Previous studies revealed limited molecular mechanisms of FLNB-related diseases but most of them were carried out with HEK293 cells from the kidney which could not reproduce FLNB's specificity to skeletal tissues. Instead, we elected to use ATDC5, a chondrogenic stem cell line widely used to study endochondral osteogenesis. In this study, we established FLNB-transfected ATDC5 cell model. We reported a pedigree of LS with mutation of FLNBG1586R and reviewed a case of BD with mutation of FLNBL171R . Using the ATDC5 cell model above, we compared cellular and molecular phenotypes of BD-associated FLNBL171R and LS-associated FLNBG1586R . We found that while both phenotypes had an increased expression of Runx2, FLNBL171R-expressing ATDC5 cells presented globular aggregation of FLNB protein and increased cellular apoptosis rate while FLNBG1586R-expressing ATDC5 cells presented evenly distributed FLNB protein and decreased cellular migration. These findings support our explanation for the cause of differences in clinical phenotypes between LS and BD. Our study makes a comparative analysis of two extremes of the FLNB-mutated autosomal dominant spectrum, relating known clinical phenotypes to our new cellular and molecular findings. These results indicated next steps for future research on the role of FLNB in the physiological process of endochondral osteogenesis.
Publicações recentes
Cell-Dependent Pathogenic Roles of Filamin B in Different Skeletal Malformations.
Deciphering the Role of Filamin B Calponin-Homology Domain in Causing the Larsen Syndrome, Boomerang Dysplasia, and Atelosteogenesis Type I Spectrum Disorders via a Computational Approach.
Fetal magnetic resonance imaging of skeletal dysplasias.
Comparative analysis of the two extremes of FLNB-mutated autosomal dominant disease spectrum: from clinical phenotypes to cellular and molecular findings.
📚 EuropePMC13 artigos no totalmostrando 7
Cell-Dependent Pathogenic Roles of Filamin B in Different Skeletal Malformations.
Oxidative medicine and cellular longevityDeciphering the Role of Filamin B Calponin-Homology Domain in Causing the Larsen Syndrome, Boomerang Dysplasia, and Atelosteogenesis Type I Spectrum Disorders via a Computational Approach.
Molecules (Basel, Switzerland)Fetal magnetic resonance imaging of skeletal dysplasias.
Pediatric radiologyComparative analysis of the two extremes of FLNB-mutated autosomal dominant disease spectrum: from clinical phenotypes to cellular and molecular findings.
American journal of translational researchPiepkorn type of osteochondrodysplasia: Defining the severe end of FLNB-related skeletal disorders in three fetuses and a 106-year-old exhibit.
American journal of medical genetics. Part ASeven additional families with spondylocarpotarsal synostosis syndrome with novel biallelic deleterious variants in FLNB.
Clinical geneticsFilamin B: The next hotspot in skeletal research?
Journal of genetics and genomics = Yi chuan xue baoAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Displasia boomerang.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Displasia boomerang
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Cell-Dependent Pathogenic Roles of Filamin B in Different Skeletal Malformations.
- Deciphering the Role of Filamin B Calponin-Homology Domain in Causing the Larsen Syndrome, Boomerang Dysplasia, and Atelosteogenesis Type I Spectrum Disorders via a Computational Approach.
- Fetal magnetic resonance imaging of skeletal dysplasias.
- Seven additional families with spondylocarpotarsal synostosis syndrome with novel biallelic deleterious variants in FLNB.
- Comparative analysis of the two extremes of FLNB-mutated autosomal dominant disease spectrum: from clinical phenotypes to cellular and molecular findings.
- FLNB-Related Disorders.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1263(Orphanet)
- OMIM OMIM:112310(OMIM)
- MONDO:0007208(MONDO)
- GARD:933(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q4943512(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
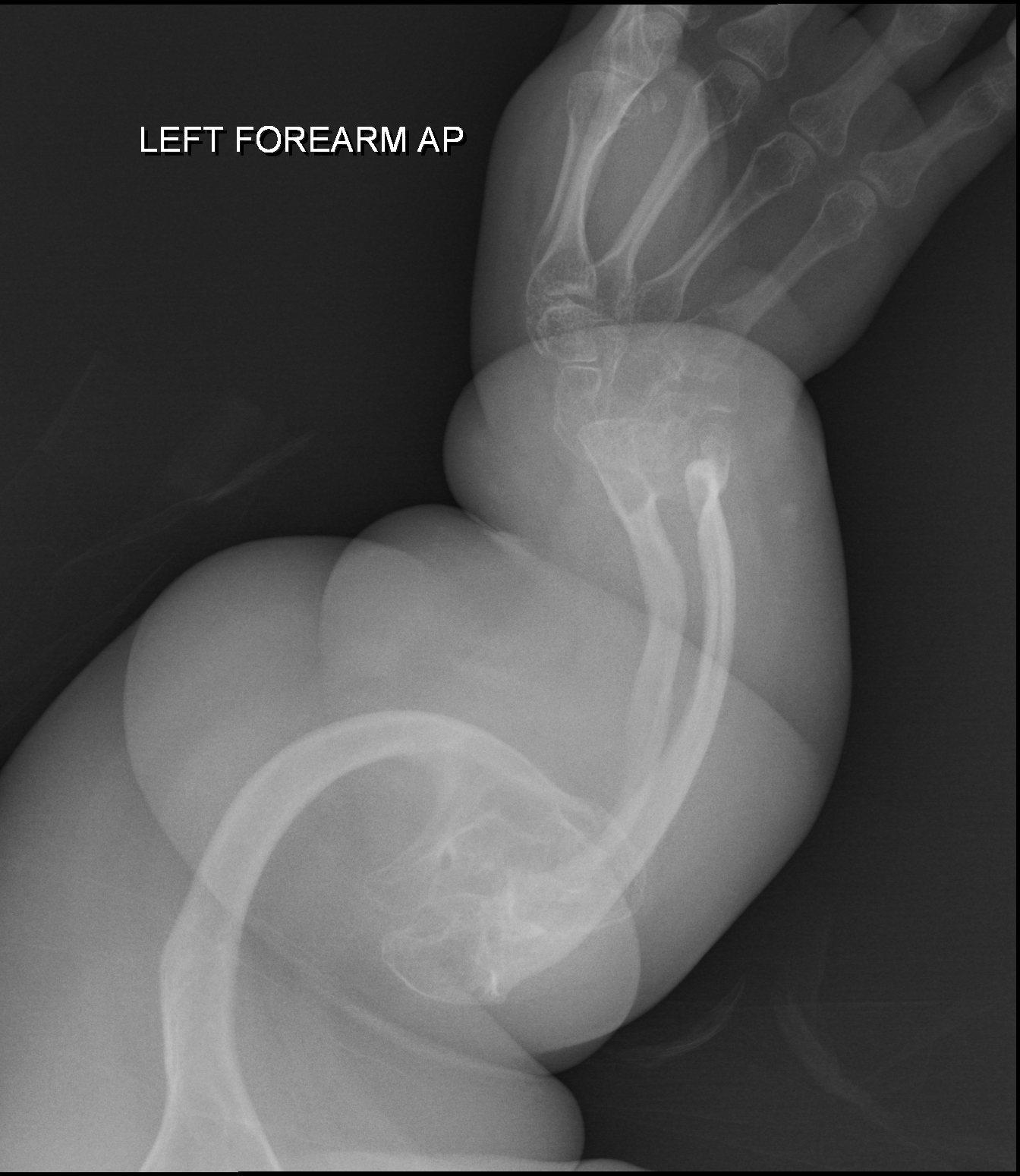
Displasia boomerang
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata