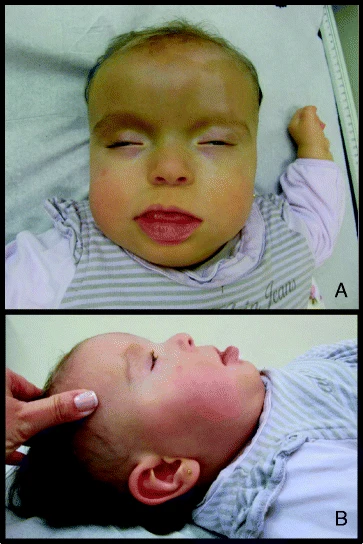
Síndrome dismórfica/anomalias congênitas múltiplas raras devido a mutações no gene CHD4. É caracterizada por atraso no desenvolvimento, atraso na fala e grau variável de deficiência intelectual (principalmente média a moderada, mas alguns pacientes também podem ter inteligência normal). Embora as manifestações clínicas sejam significativamente variáveis entre os pacientes, a maioria dos pacientes manifesta características faciais dismórficas (às vezes podem incluir macrocefalia), defeitos cardíacos congênitos, hipotonia e anormalidades oftalmológicas. Outras características clínicas podem incluir anomalias estruturais cerebrais, anomalias esqueléticas, deficiência auditiva e hipogonadismo (apenas em homens).
Introdução
O que você precisa saber de cara
Síndrome dismórfica/anomalias congênitas múltiplas raras devido a mutações no gene CHD4. É caracterizada por atraso no desenvolvimento, atraso na fala e grau variável de deficiência intelectual (principalmente média a moderada, mas alguns pacientes também podem ter inteligência normal). Embora as manifestações clínicas sejam significativamente variáveis entre os pacientes, a maioria dos pacientes manifesta características faciais dismórficas (às vezes podem incluir macrocefalia), defeitos cardíacos congênitos, hipotonia e anormalidades oftalmológicas. Outras características clínicas podem incluir anomalias estruturais cerebrais, anomalias esqueléticas, deficiência auditiva e hipogonadismo (apenas em homens).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 17 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 40 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisATP-dependent chromatin-remodeling factor that binds and distorts nucleosomal DNA (PubMed:28977666, PubMed:32543371). Acts as a component of the histone deacetylase NuRD complex which participates in the remodeling of chromatin (PubMed:16428440, PubMed:17626165, PubMed:28977666, PubMed:9804427). Localizes to acetylated damaged chromatin in a ZMYND8-dependent manner, to promote transcriptional repression and double-strand break repair by homologous recombination (PubMed:25593309). Involved in neu
NucleusCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Sifrim-Hitz-Weiss syndrome
An autosomal dominant syndrome characterized by intellectual disability, variable congenital defects affecting cardiac, skeletal, and urogenital systems. Short stature, macrocephaly, hearing impairment, and facial dysmorphism are present in some patients.
Variantes genéticas (ClinVar)
277 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
10 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença do neurodesenvolvimento CHD4-relacionada
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Discovery of a DNA methylation profile in individuals with Sifrim-Hitz-Weiss syndrome.
Pathogenic heterozygous variants in CHD4 cause Sifrim-Hitz-Weiss syndrome, a neurodevelopmental disorder associated with brain anomalies, heart defects, macrocephaly, hypogonadism, and additional features with variable expressivity. Most individuals have non-recurrent missense variants, complicating variant interpretation. A few were reported with truncating variants, and their role in disease is unclear. DNA methylation episignatures have emerged as highly accurate diagnostic biomarkers in a growing number of rare diseases. We aimed to study evidence for the existence of a CHD4-related DNA methylation episignature. We collected blood DNA samples and/or clinical information from 39 individuals with CHD4 variants, including missense and truncating variants. Genomic DNA methylation analysis was performed on 28 samples. We identified a sensitive and specific DNA methylation episignature in samples with pathogenic missense variants within the ATPase/helicase domain. The same episignature was observed in a family with variable expressivity, a de novo variant near the PHD domain, variants of uncertain significance within the ATPase/helicase domain, and a sample with compound heterozygous variants. DNA methylation data revealed higher percentages of shared probes with BAFopathies, CHD8, and the terminal ADNP variants encoding a protein known to form the ChAHP complex with CHD4. Truncating variants, as well as a sample with a recurrent pathogenic missense variant, exhibited DNA methylation profiles distinct from the ATPase/helicase domain episignature. These DNA methylation differences, together with the distinct clinical features observed in those individuals, provide preliminary evidence for clinical and molecular sub-types in the CHD4-related disorder.
The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis.
Sifrim-Hitz-Weiss syndrome (SIHIWES) is a recently described multisystemic neurodevelopmental disorder caused by de novo variants inCHD4. In this study, we investigated the clinical spectrum of the disorder, genotype-phenotype correlations, and the effect of different missense variants on CHD4 function. We collected clinical and molecular data from 32 individuals with mostly de novo variants in CHD4, identified through next-generation sequencing. We performed adenosine triphosphate (ATP) hydrolysis and nucleosome remodeling assays on variants from five different CHD4 domains. The majority of participants had global developmental delay, mild to moderate intellectual disability, brain anomalies, congenital heart defects, and dysmorphic features. Macrocephaly was a frequent but not universal finding. Additional common abnormalities included hypogonadism in males, skeletal and limb anomalies, hearing impairment, and ophthalmic abnormalities. The majority of variants were nontruncating and affected the SNF2-like region of the protein. We did not identify genotype-phenotype correlations based on the type or location of variants. Alterations in ATP hydrolysis and chromatin remodeling activities were observed in variants from different domains. The CHD4-related syndrome is a multisystemic neurodevelopmental disorder. Missense substitutions in different protein domains alter CHD4 function in a variant-specific manner, but result in a similar phenotype in humans.
The NuRD complex and macrocephaly associated neurodevelopmental disorders.
The nucleosome remodeling and deacetylase (NuRD) complex is a major regulator of gene expression involved in pluripotency, lineage commitment, and corticogenesis. This important complex is composed of seven different proteins, with mutations in CHD3, CHD4, and GATAD2B being associated with neurodevelopmental disorders presenting with macrocephaly and intellectual disability similar to other overgrowth and intellectual disability (OGID) syndromes. Pathogenic variants in CHD3 and CHD4 primarily involve disruption of enzymatic function. GATAD2B variants include loss-of-function mutations that alter protein dosage and missense variants that involve either of two conserved domains (CR1 and CR2) known to interact with other NuRD proteins. In addition to macrocephaly and intellectual disability, CHD3 variants are associated with inguinal hernias and apraxia of speech; whereas CHD4 variants are associated with skeletal anomalies, deafness, and cardiac defects. GATAD2B-associated neurodevelopmental disorder (GAND) has phenotypic overlap with both of these disorders. Of note, structural models of NuRD indicate that CHD3 and CHD4 require direct contact with the GATAD2B-CR2 domain to interact with the rest of the complex. Therefore, the phenotypic overlaps of CHD3- and CHD4-related disorders with GAND are consistent with a loss in the ability of GATAD2B to recruit CHD3 or CHD4 to the complex. The shared features of these neurodevelopmental disorders may represent a new class of OGID syndrome: the NuRDopathies.
Publicações recentes
The NuRD complex and macrocephaly associated neurodevelopmental disorders.
The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis.
📚 EuropePMCmostrando 3
Discovery of a DNA methylation profile in individuals with Sifrim-Hitz-Weiss syndrome.
American journal of human geneticsThe NuRD complex and macrocephaly associated neurodevelopmental disorders.
American journal of medical genetics. Part C, Seminars in medical geneticsThe CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis.
Genetics in medicine : official journal of the American College of Medical GeneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença do neurodesenvolvimento CHD4-relacionada.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença do neurodesenvolvimento CHD4-relacionada
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Discovery of a DNA methylation profile in individuals with Sifrim-Hitz-Weiss syndrome.
- The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis.Genetics in medicine : official journal of the American College of Medical Genetics· 2020· PMID 31388190mais citado
- The NuRD complex and macrocephaly associated neurodevelopmental disorders.American journal of medical genetics. Part C, Seminars in medical genetics· 2019· PMID 31737996mais citado
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:653712(Orphanet)
- OMIM OMIM:617159(OMIM)
- MONDO:0014946(MONDO)
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Doença do neurodesenvolvimento CHD4-relacionada
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS