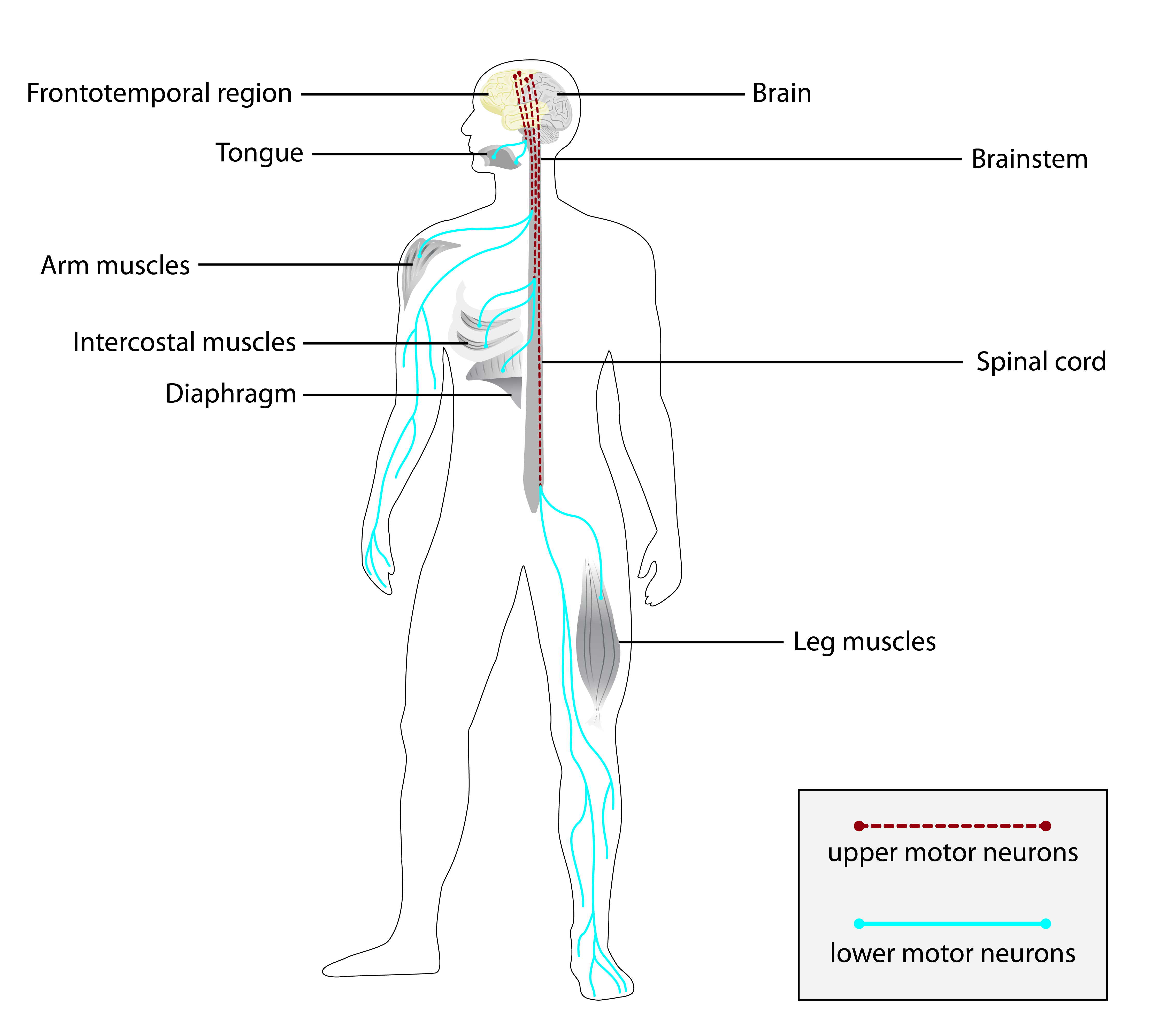
A miopatia distal de início na idade adulta devido à mutação VCP é um distúrbio genético raro de miopatia distal, caracterizado por fraqueza muscular distal da perna de início na meia-idade, atrofia no compartimento anterior resultando em queda do pé, sem fraqueza muscular esquelética proximal ou escapular. Foram relatadas demência rapidamente progressiva, doença óssea de Paget e fraqueza nas mãos. A biópsia muscular mostra alterações miopáticas pronunciadas com vacúolos orlados.
Introdução
O que você precisa saber de cara
A miopatia distal de início na idade adulta devido à mutação VCP é um distúrbio genético raro de miopatia distal, caracterizado por fraqueza muscular distal da perna de início na meia-idade, atrofia no compartimento anterior resultando em queda do pé, sem fraqueza muscular esquelética proximal ou escapular. Foram relatadas demência rapidamente progressiva, doença óssea de Paget e fraqueza nas mãos. A biópsia muscular mostra alterações miopáticas pronunciadas com vacúolos orlados.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 10 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 31 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Necessary for the fragmentation of Golgi stacks during mitosis and for their reassembly after mitosis. Involved in the formation of the transitional endoplasmic reticulum (tER). The transfer of membranes from the endoplasmic reticulum to the Golgi apparatus occurs via 50-70 nm transition vesicles which derive from part-rough, part-smooth transitional elements of the endoplasmic reticulum (tER). Vesicle budding from the tER is an ATP-dependent process. The ternary complex containing UFD1, VCP and
Cytoplasm, cytosolEndoplasmic reticulumNucleusCytoplasm, Stress granule
Inclusion body myopathy with early-onset Paget disease with or without frontotemporal dementia 1
An autosomal dominant disease characterized by disabling muscle weakness clinically resembling to limb girdle muscular dystrophy, osteolytic bone lesions consistent with Paget disease, and premature frontotemporal dementia. Clinical features show incomplete penetrance.
Variantes genéticas (ClinVar)
206 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
21 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Miopatia distal por mutação VCP com início no adulto
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Phenotypic diversity in an international Cure VCP Disease registry.
Dominant mutations in valosin-containing protein (VCP) gene cause an adult onset inclusion body myopathy, Paget's disease of bone, and frontotemporal dementia also termed multisystem proteinopathy (MSP). The genotype-phenotype relationships in VCP-related MSP are still being defined; in order to understand this better, we investigated the phenotypic diversity and patterns of weakness in the Cure VCP Disease Patient Registry. Cure VCP Disease, Inc. was founded in 2018 for the purpose of connecting patients with VCP gene mutations and researchers to help advance treatments and cures. Cure VCP Disease Patient Registry is maintained by Coordination of Rare Diseases at Sanford. The results of two questionnaires with a 5-point Likert scale questions regarding to patients' disease onset, symptoms, and daily life were obtained from 59 participants (28 males and 31 females) between June 2018 and May 2020. Independent of the registry, 22 patients were examined at the Cure VCP Disease annual patient conference in 2019. In the questionnaires of the registry, fifty-three patients (90%) reported that they were with inclusion body myopathy, 17 patients (29%) with Paget's disease of bone, eight patients (14%) with dementia, two patients (3%) with amyotrophic lateral sclerosis, and a patient with parkinsonism. Thirteen patients (22%) reported dysphagia and 25 patients (42%) reported dyspnea on exertion. A self-reported functional rating scale for motor function identified challenges with sit to stand (72%), walking (67%), and climbing stairs (85%). Thirty-five (59%) patients in the registry answered that their quality of life is more than good. As for the weakness pattern of the 22 patients who were evaluated at the Cure VCP Disease annual conference, 50% of patients had facial weakness, 55% had scapular winging, 68% had upper proximal weakness, 41% had upper distal weakness, 77% had lower proximal, and 64% had lower distal weakness. The Cure VCP Disease Patient Registry is useful for deepening the understanding of patient daily life, which would be a basis to develop appropriate clinical outcome measures. The registry data is consistent with previous studies evaluating VCP patients in the clinical setting. Patient advocacy groups are essential in developing and maintaining disease registries.
Novel valosin-containing protein mutations associated with multisystem proteinopathy.
Over fifty missense mutations in the gene coding for valosin-containing protein (VCP) are associated with a unique autosomal dominant adult-onset progressive disease associated with combinations of proximo-distal inclusion body myopathy (IBM), Paget's disease of bone (PDB), frontotemporal dementia (FTD), and amyotrophic lateral sclerosis (ALS). We report the clinical, histological, and molecular findings in four new patients/families carrying novel VCP mutations: c.474 G > A (p.M158I); c.478 G > C (p.A160P); c.383G > C (p.G128A); and c.382G > T (p.G128C). Clinical features included myopathy, PDB, ALS and Parkinson's disease though frontotemporal dementia was not an associated feature in these families. One of the patients was noted to have severe manifestations of PDB and was suspected of having neoplasia. There were wide inter- and intra-familial variations making genotype-phenotype correlations difficult between the novel mutations and frequency or age of onset of IBM, PDB, FTD, ALS and Parkinson's disease. Increasing awareness of the full spectrum of clinical presentations will improve diagnosis of VCP-related diseases and thus proactively manage or prevent associated clinical features such as PDB.
Publicações recentes
Phenotypic diversity in an international Cure VCP Disease registry.
Novel valosin-containing protein mutations associated with multisystem proteinopathy.
🥉 Relato de casoCharacterization of the Asian myopathy patients with VCP mutations.
Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia.
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Miopatia distal por mutação VCP com início no adulto.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Miopatia distal por mutação VCP com início no adulto
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Phenotypic diversity in an international Cure VCP Disease registry.
- Novel valosin-containing protein mutations associated with multisystem proteinopathy.
- Characterization of the Asian myopathy patients with VCP mutations.
- Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:329478(Orphanet)
- MONDO:0018006(MONDO)
- GARD:21492(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55787678(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Miopatia distal por mutação VCP com início no adulto
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata