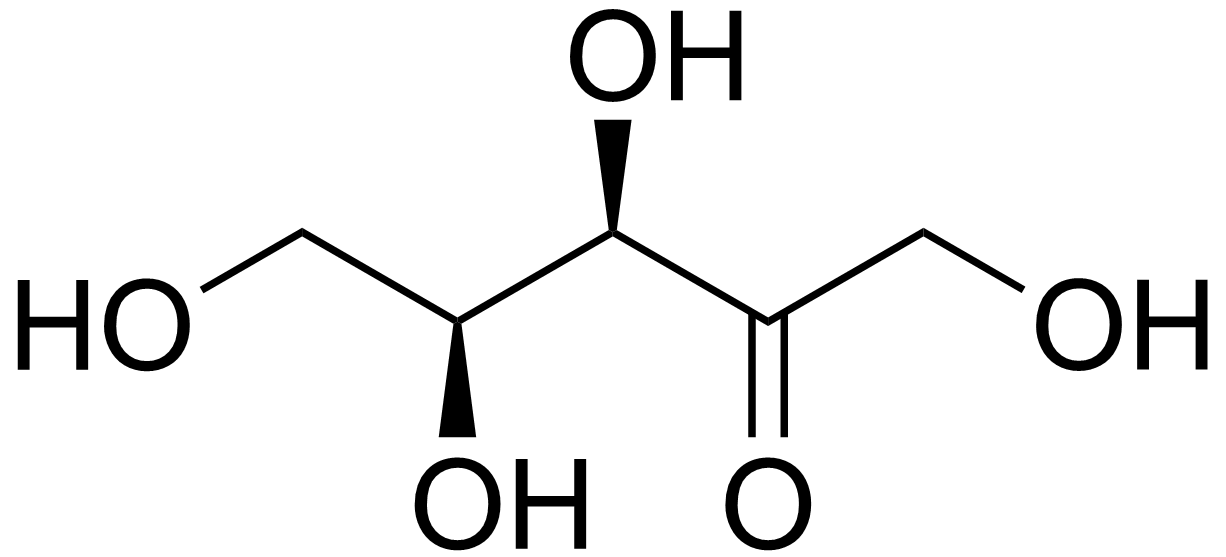
A pentosúria é um erro inato do metabolismo que se caracteriza pela excreção de 1 a 4 g da pentose L-xilulose na urina por dia.
Introdução
O que você precisa saber de cara
A pentosúria é um erro inato do metabolismo que se caracteriza pela excreção de 1 a 4 g da pentose L-xilulose na urina por dia.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 5 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Catalyzes the NADPH-dependent reduction of several pentoses, tetroses, trioses, alpha-dicarbonyl compounds and L-xylulose (PubMed:11882650, PubMed:19337691, PubMed:40737316). Can use both NAD and NADP as cosubstrate but shows higher activity with NADP (PubMed:11882650). Participates in the uronate cycle of glucose metabolism (PubMed:11882650). May play a role in the water absorption and cellular osmoregulation in the proximal renal tubules by producing xylitol, an osmolyte, thereby preventing os
Apical cell membraneCytoplasmic vesicle, secretory vesicle, acrosome
Pentosuria
An inborn error of metabolism characterized by excessive urinary excretion of L-xylulose.
Variantes genéticas (ClinVar)
23 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 2 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Pentosúria
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
C11orf54 catalyzes L-xylulose formation in human metabolism.
Excretion of L-xylulose is the hallmark of pentosuria, the fourth of Garrod's inborn errors of metabolism, yet the molecular basis for L-xylulose formation remains unknown. Here, by projecting coevolutionary data for 511,114 orthogroups across 1,929 eukaryotic genomes onto metabolic maps, we screen for unmapped genes in human metabolism. Among these, we show that the DUF1907 domain of C11orf54 catalyzes formation of L-xylulose by establishing a zinc-coordinated Michaelis complex with β-keto-L-gulonate (BKG). The identification of BKG decarboxylase completes the pentose pathway, in which pentose sugars are produced by decarboxylation of nonphosphorylated hexose precursors. The pathway was present in the unicellular ancestor of animals and is conserved in all deuterostomes, in contrast to the alternative L-ascorbate (vitamin C) biosynthesis pathway. An increased flux toward pentoses may have represented an evolutionary tradeoff, favoring energy metabolism and redox cofactor balance at the expense of ascorbate biosynthesis in organisms, such as humans and other Haplorhini primates, where dietary vitamin C intake prevents scurvy.
A Feeling for the Human Subject: Margaret Lasker and the Genetic Puzzle of Pentosuria.
In 1933 Margaret Lasker, a biochemist who worked at the labs of Montefiore Hospital in New York, developed an accurate method for the differentiation between pentosuria and diabetes. Research into pentosuria, and mostly its genetic aspects, became Lasker's lifelong passion. Since research was not part of her job description, she conducted the chief part of her study in her home kitchen. Lasker's extensive and personal correspondence with her patients and their families may be the secret key for her success in maintaining a prolonged research career against all odds. Laker's last article was published in 1955 in Human Biology, presenting data on 72 cases of pentosuria, which occurs almost exclusively in Ashkenazi Jews. More than half a century later, and long after Lasker was gone, her well kept data and family records allowed the discovery of two mutations in the DCXR gene, by Mary-Claire King and her team.
Milestones in treatments for inborn errors of metabolism: Reflections on Where chemistry and medicine meet.
From Sir Archibald Garrod's initial description of the tetrad of albinism, alkaptonuria, cystinuria, and pentosuria to today, the field of medicine dedicated to inborn errors of metabolism has evolved from disease identification and mechanistic discovery to the development of therapies designed to subvert biochemical defects. In this review, we highlight major milestones in the treatment and diagnosis of inborn errors of metabolism, starting with dietary therapy for phenylketonuria in the 1950s and 1960s, and ending with current approaches in genetic manipulation.
Inborn errors of metabolism in the 21st century: past to present.
The 21st century is an exciting time to be in the field of metabolic medicine. As with many fields, one of the keys to anticipating the future is to understand the past. The term "inborn error of metabolism" was first coined in 1908 by Sir Archibald Garrod, in reference to four disorders (alkaptonuria, pentosuria, cystinuria and albinism). The first (and still most definitive) textbook on the subject, "The Metabolic Basis of Inherited Disease" was initially published in 1960 and covered 80 disorders in 1,477 pages. After the eighth edition of this text became unwieldy at 6,338 pages in 4 volumes covering more than 1,000 disorders, the book was changed to an online reference text with 259 chapters and is still growing. Current newborn screening on a few dried blood spots on filter paper identifies more than 1 in 2,000 newborns as having a metabolic disorder. The availability of metabolomic and genomic analyses is resulting in the diagnosis of many new disorders. Enzyme replacement therapy (ERT) has provided treatments for previously untreatable metabolic disorders, and the promise of gene therapy on the near horizon will certainly revolutionize the field.
Publicações recentes
C11orf54 catalyzes L-xylulose formation in human metabolism.
A Feeling for the Human Subject: Margaret Lasker and the Genetic Puzzle of Pentosuria.
Milestones in treatments for inborn errors of metabolism: Reflections on Where chemistry and medicine meet.
Inborn errors of metabolism in the 21(st) century: past to present.
Human DCXR - another 'moonlighting protein' involved in sugar metabolism, carbonyl detoxification, cell adhesion and male fertility?
📚 EuropePMC47 artigos no totalmostrando 4
C11orf54 catalyzes L-xylulose formation in human metabolism.
Proceedings of the National Academy of Sciences of the United States of AmericaA Feeling for the Human Subject: Margaret Lasker and the Genetic Puzzle of Pentosuria.
Journal of the history of biologyMilestones in treatments for inborn errors of metabolism: Reflections on Where chemistry and medicine meet.
American journal of medical genetics. Part AInborn errors of metabolism in the 21st century: past to present.
Annals of translational medicineAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Pentosúria.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Pentosúria
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- C11orf54 catalyzes L-xylulose formation in human metabolism.Proceedings of the National Academy of Sciences of the United States of America· 2025· PMID 40737316mais citado
- A Feeling for the Human Subject: Margaret Lasker and the Genetic Puzzle of Pentosuria.
- Milestones in treatments for inborn errors of metabolism: Reflections on Where chemistry and medicine meet.
- Inborn errors of metabolism in the 21st century: past to present.
- Human DCXR - another 'moonlighting protein' involved in sugar metabolism, carbonyl detoxification, cell adhesion and male fertility?
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2843(Orphanet)
- OMIM OMIM:260800(OMIM)
- MONDO:0009846(MONDO)
- GARD:418(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q1965082(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Pentosúria
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata