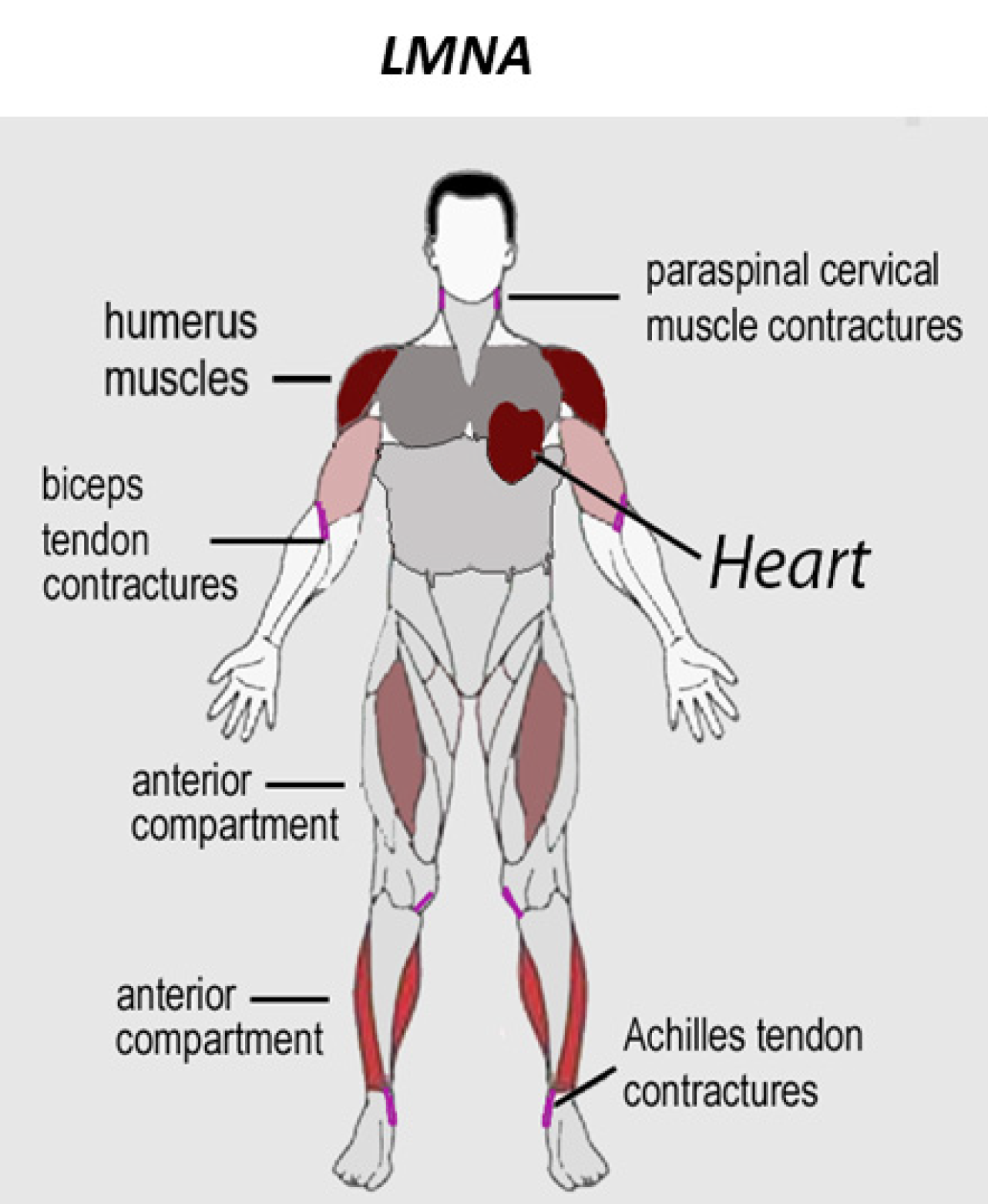
A distrofia muscular de Emery-Dreifuss (EDMD) é caracterizada por fraqueza e atrofia muscular, com contraturas articulares precoces e cardiomiopatia.
Introdução
O que você precisa saber de cara
A distrofia muscular de Emery-Dreifuss (EDMD) é caracterizada por fraqueza e atrofia muscular, com contraturas articulares precoces e cardiomiopatia.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 36 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 128 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
7 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, X-linked recessive.
May have an involvement in muscle development or hypertrophy
CytoplasmNucleusCytoplasm, cytosol
Emery-Dreifuss muscular dystrophy 6, X-linked
A form of Emery-Dreifuss muscular dystrophy, a degenerative myopathy characterized by weakness and atrophy of muscle without involvement of the nervous system, early contractures of the elbows, Achilles tendons and spine, and cardiomyopathy associated with cardiac conduction defects.
Multi-isomeric modular protein which forms a linking network between organelles and the actin cytoskeleton to maintain the subcellular spatial organization. As a component of the LINC (LInker of Nucleoskeleton and Cytoskeleton) complex involved in the connection between the nuclear lamina and the cytoskeleton. The nucleocytoplasmic interactions established by the LINC complex play an important role in the transmission of mechanical forces across the nuclear envelope and in nuclear movement and p
Nucleus outer membraneSarcoplasmic reticulum membraneCell membraneCytoplasm, cytoskeletonMitochondrionNucleus, nucleoplasmCytoplasm, myofibril, sarcomere, Z lineCell junction, focal adhesion
Emery-Dreifuss muscular dystrophy 5, autosomal dominant
A form of Emery-Dreifuss muscular dystrophy, a degenerative myopathy characterized by weakness and atrophy of muscle without involvement of the nervous system, early contractures of the elbows, Achilles tendons and spine, and cardiomyopathy associated with cardiac conduction defects.
Lamins are intermediate filament proteins that assemble into a filamentous meshwork, and which constitute the major components of the nuclear lamina, a fibrous layer on the nucleoplasmic side of the inner nuclear membrane (PubMed:10080180, PubMed:10580070, PubMed:10587585, PubMed:10814726, PubMed:11799477, PubMed:12075506, PubMed:12927431, PubMed:15317753, PubMed:18551513, PubMed:18611980, PubMed:2188730, PubMed:22431096, PubMed:2344612, PubMed:23666920, PubMed:24741066, PubMed:31434876, PubMed:
Nucleus laminaNucleus envelopeNucleus, nucleoplasmNucleus matrixNucleus speckle
Emery-Dreifuss muscular dystrophy 2, autosomal dominant
A form of Emery-Dreifuss muscular dystrophy, a degenerative myopathy characterized by weakness and atrophy of muscle without involvement of the nervous system, early contractures of the elbows, Achilles tendons and spine, and cardiomyopathy associated with cardiac conduction defects.
Multi-isomeric modular protein which forms a linking network between organelles and the actin cytoskeleton to maintain the subcellular spatial organization. As a component of the LINC (LInker of Nucleoskeleton and Cytoskeleton) complex involved in the connection between the nuclear lamina and the cytoskeleton. The nucleocytoplasmic interactions established by the LINC complex play an important role in the transmission of mechanical forces across the nuclear envelope and in nuclear movement and p
Nucleus outer membraneNucleusNucleus envelopeCytoplasm, cytoskeletonCytoplasm, myofibril, sarcomereGolgi apparatus
Spinocerebellar ataxia, autosomal recessive, 8
A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAR8 is an autosomal recessive form.
Myosins are actin-based motor molecules with ATPase activity essential for muscle contraction. Forms regular bipolar thick filaments that, together with actin thin filaments, constitute the fundamental contractile unit of skeletal and cardiac muscle
Cytoplasm, myofibrilCytoplasm, myofibril, sarcomere
Cardiomyopathy, familial hypertrophic, 1
A hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death.
Stabilizes and promotes the formation of a nuclear actin cortical network. Stimulates actin polymerization in vitro by binding and stabilizing the pointed end of growing filaments. Inhibits beta-catenin activity by preventing its accumulation in the nucleus. Acts by influencing the nuclear accumulation of beta-catenin through a CRM1-dependent export pathway. Links centrosomes to the nuclear envelope via a microtubule association. Required for proper localization of non-farnesylated prelamin-A/C.
Nucleus inner membraneNucleus outer membrane
Emery-Dreifuss muscular dystrophy 1, X-linked
A form of Emery-Dreifuss muscular dystrophy, a degenerative myopathy characterized by weakness and atrophy of muscle without involvement of the nervous system, early contractures of the elbows, Achilles tendons and spine, and cardiomyopathy associated with cardiac conduction defects.
May have an important role in maintaining nuclear envelope structure by organizing protein complexes at the inner nuclear membrane. Required for retaining emerin at the inner nuclear membrane (By similarity). Plays a role in the modulation of innate immune signaling through the cGAS-STING pathway by interacting with RNF26 (PubMed:32614325). In addition, functions as a critical signaling component in mediating NF-kappa-B activation by acting downstream of EGFR and upstream of CARD10 (PubMed:27991
Endoplasmic reticulum membraneNucleus inner membraneCell membrane
Arrhythmogenic right ventricular dysplasia, familial, 5
A congenital heart disease characterized by infiltration of adipose and fibrous tissue into the right ventricle and loss of myocardial cells, resulting in ventricular and supraventricular arrhythmias.
Variantes genéticas (ClinVar)
1.423 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 10.747 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
15 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Distrofia muscular de Emery-Dreifuss
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
2 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Mostrando amostra de 18 publicações de um total de 286
Dominant myosin storage myopathy mutations disrupt striated muscles in Drosophila and the myosin tail-tail interactome of human cardiac thick filaments.
Myosin storage myopathy (MSM) is a rare skeletal muscle disorder caused by mutations in the slow muscle/β-cardiac myosin heavy chain (MHC) gene. MSM missense mutations frequently disrupt the tail's stabilizing heptad repeat motif. Disease hallmarks include subsarcolemmal hyaline-like β-MHC aggregates, muscle weakness, and, occasionally, cardiomyopathy. We generated transgenic, heterozygous Drosophila to examine the dominant physiological and structural effects of the L1793P, R1845W, and E1883K MHC MSM mutations on diverse muscles. The MHC variants reduced lifespan and flight and jump abilities. Moreover, confocal and electron microscopy revealed that they provoked indirect flight muscle breaks and myofibrillar disarray/degeneration with filamentous inclusions. Incorporation of GFP-myosin enabled in situ determination of thick filament lengths, which were significantly reduced in all mutants. Semiautomated heartbeat analysis uncovered aberrant cardiac function, which worsened with age. Thus, our fly models phenocopied traits observed among MSM patients. We additionally mapped the mutations onto a recently determined, 6 Å resolution, cryo-EM structure of the human cardiac thick filament. The R1845W mutation replaces a basic arginine with a polar-neutral, bulkier tryptophan, while E1883K reverses charge at critical filament loci. Both would be expected to disrupt the core and the outer shell of the backbone structure. Replacing L1793 with a proline, a potent breaker of α-helices, could disturb the coiled-coil of the myosin rod and alter the tail-tail interactome. Hence, all mutations likely destabilize and weaken the filament backbone. This may trigger disease in humans, while potentially analogous perturbations are likely to yield the observed thick filament and muscle disruption in our fly models.
A novel heterozygous missense MYH7 mutation potentially causes an autosomal dominant form of myosin storage myopathy with dilated cardiomyopathy.
The MYH7 gene, which encodes the slow/ß-cardiac myosin heavy chain, is mutated in myosin storage myopathy (MSM). The clinical spectrum of MSM is quite heterogeneous in that it ranges from cardiomyopathies to skeletal myopathies or a combination of both, depending on the affected region. In this study, we performed clinical and molecular examinations of the proband of an Iranian family with MSM in an autosomal dominant condition exhibiting proximal muscle weakness and dilated cardiomyopathy. Following thorough clinical and paraclinical examinations, whole-exome sequencing `was performed on the proband (II-5). Pathogenicity prediction of the candidate variant was performed through in-silico analysis. Co-segregation analysis of the WES data among the family members was carried out by PCR-based Sanger sequencing. A novel heterozygous missense variant, MYH7 (NM_000257): c.C1888A: p.Pro630Thr, was found in the DNA of the proband and his children and confirmed by Sanger sequencing. The in-silico analysis revealed that p.Pro630Thr substitution was deleterious. The novel sequence variant fell within a highly conserved region of the head domain. Our findings expand the spectrum of MYH7 mutations. This finding could improve genetic counseling and prenatal diagnosis in families with clinical manifestations associated with MYH7-related myopathy.
Cardiac Rehabilitation in a Transplanted Person with Emery-Dreifuss Muscular Dystrophy.
Emery-Dreifuss muscular dystrophy is a rare hereditary neuromuscular disease. Its manifestations begin primarily in childhood. The most frequent manifestations are progressive muscle weakness, atrophy that usually begins in the scapula-vertebral region, extending later to the pelvic girdle, and spinal stiffness. Patients can also manifest cardiac involvement as palpitations, syncope, exercise intolerance, congestive heart failure, and variable heart rhythm disturbances. 1 - 3 The presence and severity of these manifestations can vary according to the individual and the disease's subtypes. 2 Cardiac involvement is the most worrisome feature of this disease, and there are some reports of the need for heart transplantation in this dystrophy. 4. A distrofia muscular de Emery-Dreifuss é uma doença neuromuscular hereditária rara. Suas manifestações começam principalmente na infância. As manifestações mais frequentes são fraqueza muscular progressiva, atrofia que geralmente se inicia na região escápulo-vertebral, estendendo-se posteriormente para a cintura pélvica e rigidez da coluna vertebral. Os pacientes também podem manifestar envolvimento cardíaco como palpitações, síncope, intolerância ao exercício, insuficiência cardíaca congestiva e distúrbios variáveis do ritmo cardíaco. 1 - 3 A presença e a gravidade dessas manifestações podem variar de acordo com o indivíduo e os subtipos da doença. 2 O envolvimento cardíaco é a característica mais preocupante desta doença, havendo alguns relatos da necessidade de transplante cardíaco nesta distrofia. 4. Emery-Dreifuss muscular dystrophy is a rare hereditary neuromuscular disease. Its manifestations begin primarily in childhood. The most frequent manifestations are progressive muscle weakness, atrophy that usually begins in the scapula-vertebral region, extending later to the pelvic girdle, and spinal stiffness. Patients can also manifest cardiac involvement as palpitations, syncope, exercise intolerance, congestive heart failure, and variable heart rhythm disturbances. - The presence and severity of these manifestations can vary according to the individual and the disease’s subtypes. Cardiac involvement is the most worrisome feature of this disease, and there are some reports of the need for heart transplantation in this dystrophy.
Identification of novel FHL1 mutations associated with X-linked scapuloperoneal myopathy in unrelated Chinese patients.
Mutations in the FHL1 gene can be associated with a variety of X-linked myopathies and cardiomyopathies, among which X-linked dominant scapuloperoneal myopathy is a rare phenotype. We collected the clinical data of two unrelated Chinese patients with X-linked scapuloperoneal myopathy and analyzed their clinical, pathological, muscle imaging, and genetic features. Both patients were characterized by scapular winging, bilateral Achilles tendon contractures, and weakness in shoulder-girdle and peroneal muscles. Muscle biopsy revealed myopathic changes, and no reducing bodies were found. Muscle magnetic resonance imaging was dominated by fatty infiltration, with minor edema-like findings. Genetic analysis revealed two novel mutations in the FHL1 gene: c.380T > C (p.F127S) and c.802C > T (p.Q268*), which were located in the LIM2 domain and the C-terminal sequence, respectively. To our knowledge, this is the first report of X-linked scapuloperoneal myopathy in the Chinese population. Our findings broadened the genetic and ethnic spectrum of FHL1-related disorders and proposed to look for variants in the FHL1 gene when scapuloperoneal myopathy is observed in the clinical work.
253rd ENMC international workshop: Striated muscle laminopathies - natural history and clinical trial readiness. 24-26 June 2022, Hoofddorp, the Netherlands.
Publicações recentes
WNT5a-Mediated Aberrant Actin Filament Dynamics Drive Cardiac Pathogenic Phenotypes in LMNA-Related Emery-Dreifuss Muscular Dystrophy.
Rats lacking emerin develop muscle pathologies and molecular alterations found in humans with X-linked EDMD.
Successful Ablation of Multifocal Atrial Flutter in Pediatric Emery-Dreifuss Muscular Dystrophy Patient Using Pulsed Field Ablation.
X-linked Emery-Dreifuss muscular dystrophy caused by a novel FHL1 mutation: A case report.
Cargo Recognition of Nesprin-2 by the Dynein Adapter Bicaudal D2 for a Nuclear Positioning Pathway That Is Important for Brain Development.
📚 EuropePMC354 artigos no totalmostrando 18
Dominant myosin storage myopathy mutations disrupt striated muscles in Drosophila and the myosin tail-tail interactome of human cardiac thick filaments.
GeneticsA novel heterozygous missense MYH7 mutation potentially causes an autosomal dominant form of myosin storage myopathy with dilated cardiomyopathy.
BMC cardiovascular disordersCardiac Rehabilitation in a Transplanted Person with Emery-Dreifuss Muscular Dystrophy.
Arquivos brasileiros de cardiologia253rd ENMC international workshop: Striated muscle laminopathies - natural history and clinical trial readiness. 24-26 June 2022, Hoofddorp, the Netherlands.
Neuromuscular disorders : NMDIdentification of novel FHL1 mutations associated with X-linked scapuloperoneal myopathy in unrelated Chinese patients.
Journal of human geneticsAn autopsy case of sudden unexpected death of a young adult with progressive intraventricular conduction delay.
Pathology, research and practiceProfile of Emery N. Brown.
Proceedings of the National Academy of Sciences of the United States of AmericaSurgical Treatment for Severe Cervical Hyperlordosis and Thoracolumar Kyphoscoliosis with Emery-Dreifuss Muscular Dystrophy: A Case Report and Literature Review.
Orthopaedic surgeryMYH7-related disorders in two Bulgarian families: Novel variants in the same region associated with different clinical manifestation and disease penetrance.
Neuromuscular disorders : NMDImpaired muscle morphology in a Drosophila model of myosin storage myopathy was supressed by overexpression of an E3 ubiquitin ligase.
Disease models & mechanismsRelationship between infrared skin radiation and muscular strength tests in patients affected by Emery-Dreifuss muscular dystrophy.
Medical hypothesesA novel missense mutation in the MYH7 gene causes an uncharacteristic phenotype of myosin storage myopathy: a case report.
BMC medical geneticsResearch progress of myosin heavy chain genes in human genetic diseases.
Yi chuan = HereditasMyosin storage myopathy mutations yield defective myosin filament assembly in vitro and disrupted myofibrillar structure and function in vivo.
Human molecular geneticsNovel phenotypic variant in the MYH7 spectrum due to a stop-loss mutation in the C-terminal region: a case report.
BMC medical geneticsMyosin Storage Myopathy in C. elegans and Human Cultured Muscle Cells.
PloS one[Application of targeted capture technology and next generation sequencing in molecular diagnosis of inherited myopathy].
Zhonghua er ke za zhi = Chinese journal of pediatricsHomozygous MYH7 R1820W mutation results in recessive myosin storage myopathy: scapuloperoneal and respiratory weakness with dilated cardiomyopathy.
Neuromuscular disorders : NMDAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Associação brasileira dedicada a Distrofias musculares.
Associação brasileira dedicada a Doença de Pompe.
Fundada pela geneticista Mayana Zatz.
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Distrofia muscular de Emery-Dreifuss
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Dominant myosin storage myopathy mutations disrupt striated muscles in Drosophila and the myosin tail-tail interactome of human cardiac thick filaments.
- A novel heterozygous missense MYH7 mutation potentially causes an autosomal dominant form of myosin storage myopathy with dilated cardiomyopathy.
- Cardiac Rehabilitation in a Transplanted Person with Emery-Dreifuss Muscular Dystrophy.
- Identification of novel FHL1 mutations associated with X-linked scapuloperoneal myopathy in unrelated Chinese patients.
- 253rd ENMC international workshop: Striated muscle laminopathies - natural history and clinical trial readiness. 24-26 June 2022, Hoofddorp, the Netherlands.
- WNT5a-Mediated Aberrant Actin Filament Dynamics Drive Cardiac Pathogenic Phenotypes in LMNA-Related Emery-Dreifuss Muscular Dystrophy.
- Rats lacking emerin develop muscle pathologies and molecular alterations found in humans with X-linked EDMD.
- Successful Ablation of Multifocal Atrial Flutter in Pediatric Emery-Dreifuss Muscular Dystrophy Patient Using Pulsed Field Ablation.
- X-linked Emery-Dreifuss muscular dystrophy caused by a novel FHL1 mutation: A case report.
- Cargo Recognition of Nesprin-2 by the Dynein Adapter Bicaudal D2 for a Nuclear Positioning Pathway That Is Important for Brain Development.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:261(Orphanet)
- MONDO:0016830(MONDO)
- GARD:6329(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q1335642(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Distrofia muscular de Emery-Dreifuss
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov