A hipoplasia cerebelar é caracterizada pela redução do volume cerebelar, embora a forma do cerebelo seja (quase) normal. Consiste em um grupo heterogêneo de distúrbios de malformação cerebelar que se manifestam como ataxia congênita não progressiva de início precoce, hipotonia e dificuldade de aprendizado motor.
Introdução
O que você precisa saber de cara
Visão geral
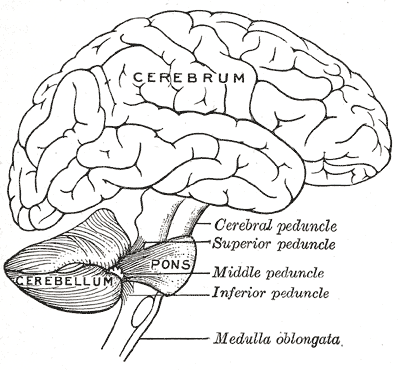
A hipoplasia pontocerebelar tipo 14 é uma doença genética rara que afeta o desenvolvimento do sistema nervoso central, especialmente o cerebelo e a ponte (estruturas localizadas na parte de trás do cérebro). A condição é herdada de forma autossômica recessiva, ou seja, ambos os pais precisam carregar uma cópia do gene alterado para que a criança manifeste a doença. A prevalência estimada é de menos de 1 caso a cada 1.000.000 de pessoas. Os primeiros sinais podem aparecer ainda antes do nascimento (período antenatal) ou logo nos primeiros dias de vida (período neonatal).[1][4]
Sinais e sintomas
Os sintomas da hipoplasia pontocerebelar tipo 14 são variados e afetam múltiplos sistemas do corpo. No sistema nervoso, podem ocorrer crises epilépticas de diferentes tipos, como crises tônico-clônicas bilaterais (que afetam os dois lados do corpo), crises mioclônicas (contrações musculares rápidas) e crises de início focal (que começam em uma área específica do cérebro). Também são comuns espasmos infantis, distonia (contrações musculares involuntárias que causam movimentos repetitivos ou posturas anormais), hipertonia (aumento do tônus muscular) e hipotonia (diminuição do tônus muscular). A mielinização do sistema nervoso central (processo de formação da bainha de mielina, que isola os neurônios) é atrasada. O desenvolvimento motor e social é atrasado, e a fala pode estar ausente. A deficiência intelectual é grave. Além disso, podem ocorrer alterações sanguíneas, como neutropenia crônica (diminuição de um tipo de glóbulo branco) e trombocitopenia (diminuição das plaquetas). Outros achados incluem hidrocefalia (acúmulo de líquido no cérebro), reflexos exaltados, tetraplegia espástica (paralisia dos quatro membros com rigidez muscular) e morte na infância.[1][4]
Causas genéticas
A hipoplasia pontocerebelar tipo 14 é causada por alterações (mutações) no gene PPIL1 (sigla em inglês para Peptidyl-prolyl cis-trans isomerase-like 1). Esse gene fornece instruções para a produção de uma proteína que participa do dobramento de outras proteínas, um processo essencial para o funcionamento adequado das células. A herança é autossômica recessiva, o que significa que a criança precisa herdar uma cópia do gene alterado de cada um dos pais para desenvolver a doença.[1][2][5]
Diagnóstico
O diagnóstico da hipoplasia pontocerebelar tipo 14 é baseado na avaliação clínica dos sinais e sintomas, em exames de imagem do sistema nervoso central (como ressonância magnética) e em testes genéticos. Os exames de imagem podem mostrar hipoplasia (desenvolvimento incompleto) do cerebelo, da ponte e do tronco cerebral, além de agenesia do corpo caloso (ausência da estrutura que conecta os dois hemisférios cerebrais) e padrão giratório simplificado (alteração na superfície do cérebro). Os testes genéticos disponíveis incluem o cariótipo com bandas G, Q ou R, a pesquisa de microdeleções/microduplicações por FISH e o sequenciamento completo do exoma (WES). Exames laboratoriais como a dosagem de alfa-fetoproteína também podem ser solicitados. Atualmente, existem 95 testes genéticos disponíveis e 25 variantes patogênicas registradas no ClinVar (banco de dados de variantes genéticas).[1][2][5]
Tratamento e manejo
Não há cura para a hipoplasia pontocerebelar tipo 14. O tratamento é sintomático e de suporte, visando controlar as crises epilépticas, a distonia e outros sintomas, além de oferecer suporte nutricional e respiratório quando necessário. O acompanhamento multidisciplinar é essencial e pode incluir neurologista, geneticista, fisioterapeuta, terapeuta ocupacional, fonoaudiólogo e psicólogo. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para o atendimento em reabilitação voltado a doenças raras. Não há medicamentos específicos aprovados para a doença, e o uso de qualquer medicação deve ser prescrito e monitorado por um médico.[1][2]
Prognóstico e qualidade de vida
O prognóstico da hipoplasia pontocerebelar tipo 14 é reservado. A doença pode levar à morte ainda na infância, devido às complicações neurológicas e sistêmicas. A qualidade de vida é significativamente afetada, com deficiência intelectual grave, ausência de fala, tetraplegia espástica e dependência total para as atividades diárias. O suporte familiar e o acesso a cuidados paliativos e de reabilitação são fundamentais para oferecer conforto e dignidade ao paciente e à família.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
A hipoplasia cerebelar é caracterizada pela redução do volume cerebelar, embora a forma do cerebelo seja (quase) normal. Consiste em um grupo heterogêneo de distúrbios de malformação cerebelar que se manifestam como ataxia congênita não progressiva de início precoce, hipotonia e dificuldade de aprendizado motor.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A hipoplasia pontocerebelar tipo 14 é uma doença genética rara que afeta o desenvolvimento do sistema nervoso central, especialmente o cerebelo e a ponte (estruturas localizadas na parte de trás do cérebro). A condição é herdada de forma autossômica recessiva, ou seja, ambos os pais precisam carregar uma cópia do gene alterado para que a criança manifeste a doença. A prevalência estimada é de menos de 1 caso a cada 1.000.000 de pessoas. Os primeiros sinais podem aparecer ainda antes do nascimento (período antenatal) ou logo nos primeiros dias de vida (período neonatal).[1][4]
Sinais e sintomas
Os sintomas da hipoplasia pontocerebelar tipo 14 são variados e afetam múltiplos sistemas do corpo. No sistema nervoso, podem ocorrer crises epilépticas de diferentes tipos, como crises tônico-clônicas bilaterais (que afetam os dois lados do corpo), crises mioclônicas (contrações musculares rápidas) e crises de início focal (que começam em uma área específica do cérebro). Também são comuns espasmos infantis, distonia (contrações musculares involuntárias que causam movimentos repetitivos ou posturas anormais), hipertonia (aumento do tônus muscular) e hipotonia (diminuição do tônus muscular). A mielinização do sistema nervoso central (processo de formação da bainha de mielina, que isola os neurônios) é atrasada. O desenvolvimento motor e social é atrasado, e a fala pode estar ausente. A deficiência intelectual é grave. Além disso, podem ocorrer alterações sanguíneas, como neutropenia crônica (diminuição de um tipo de glóbulo branco) e trombocitopenia (diminuição das plaquetas). Outros achados incluem hidrocefalia (acúmulo de líquido no cérebro), reflexos exaltados, tetraplegia espástica (paralisia dos quatro membros com rigidez muscular) e morte na infância.[1][4]
Causas genéticas
A hipoplasia pontocerebelar tipo 14 é causada por alterações (mutações) no gene PPIL1 (sigla em inglês para Peptidyl-prolyl cis-trans isomerase-like 1). Esse gene fornece instruções para a produção de uma proteína que participa do dobramento de outras proteínas, um processo essencial para o funcionamento adequado das células. A herança é autossômica recessiva, o que significa que a criança precisa herdar uma cópia do gene alterado de cada um dos pais para desenvolver a doença.[1][2][5]
Diagnóstico
O diagnóstico da hipoplasia pontocerebelar tipo 14 é baseado na avaliação clínica dos sinais e sintomas, em exames de imagem do sistema nervoso central (como ressonância magnética) e em testes genéticos. Os exames de imagem podem mostrar hipoplasia (desenvolvimento incompleto) do cerebelo, da ponte e do tronco cerebral, além de agenesia do corpo caloso (ausência da estrutura que conecta os dois hemisférios cerebrais) e padrão giratório simplificado (alteração na superfície do cérebro). Os testes genéticos disponíveis incluem o cariótipo com bandas G, Q ou R, a pesquisa de microdeleções/microduplicações por FISH e o sequenciamento completo do exoma (WES). Exames laboratoriais como a dosagem de alfa-fetoproteína também podem ser solicitados. Atualmente, existem 95 testes genéticos disponíveis e 25 variantes patogênicas registradas no ClinVar (banco de dados de variantes genéticas).[1][2][5]
Tratamento e manejo
Não há cura para a hipoplasia pontocerebelar tipo 14. O tratamento é sintomático e de suporte, visando controlar as crises epilépticas, a distonia e outros sintomas, além de oferecer suporte nutricional e respiratório quando necessário. O acompanhamento multidisciplinar é essencial e pode incluir neurologista, geneticista, fisioterapeuta, terapeuta ocupacional, fonoaudiólogo e psicólogo. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para o atendimento em reabilitação voltado a doenças raras. Não há medicamentos específicos aprovados para a doença, e o uso de qualquer medicação deve ser prescrito e monitorado por um médico.[1][2]
Prognóstico e qualidade de vida
O prognóstico da hipoplasia pontocerebelar tipo 14 é reservado. A doença pode levar à morte ainda na infância, devido às complicações neurológicas e sistêmicas. A qualidade de vida é significativamente afetada, com deficiência intelectual grave, ausência de fala, tetraplegia espástica e dependência total para as atividades diárias. O suporte familiar e o acesso a cuidados paliativos e de reabilitação são fundamentais para oferecer conforto e dignidade ao paciente e à família.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 11 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 24 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisInvolved in pre-mRNA splicing as component of the spliceosome (PubMed:11991638, PubMed:28076346, PubMed:28502770, PubMed:33220177). PPIases accelerate the folding of proteins. Catalyzes the cis-trans isomerization of proline imidic peptide bonds in oligopeptides (PubMed:16595688). Catalyzes prolyl peptide bond isomerization in CDC40/PRP17 (PubMed:33220177). Plays an important role in embryonic brain development; this function is independent of its isomerase activity (PubMed:33220177)
Nucleus
Pontocerebellar hypoplasia 14
A form of pontocerebellar hypoplasia, a disorder characterized by structural defects of the pons and cerebellum, evident upon brain imaging. PCH14 is a severe autosomal recessive form characterized by progressive microcephaly, and poor or absent psychomotor development with severely impaired intellectual development apparent from birth. Other features may include hypotonia, spastic quadriplegia, and early-onset seizures. Early death may occur in some patients.
Variantes genéticas (ClinVar)
25 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 2.116 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Hipoplasia pontocerebelar tipo 14
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Mostrando amostra de 23 publicações de um total de 322
Refining the phenotypic spectrum of PNKP-related microcephaly: a study of 27 new patients.
Biallelic pathogenic variants in PNKP are associated with microcephaly and early-onset seizures (MCSZ), ataxia with oculomotor apraxia type 4 and Charcot-Marie-Tooth disease type 2B2. We describe the clinical and neuroimaging features of 27 new patients with PNKP variants. All patients presented with early-onset seizures, congenital microcephaly and intellectual disability. In addition, we compared our results with data in the literature. Twenty-five patients presented with the classic MCSZ phenotype, while two showed a more severe clinical phenotype. The brain imaging features of the 25 patients varied significantly, but widening of the frontal lobe gyri with frontal hypoplasia and prominent cerebellar folia (consistent with atrophy) could point to PNKP-related microcephaly . In contrast, the two patients with severe phenotype showed additional brain MRI features of white matter loss and pontocerebellar hypoplasia fulfilling the criteria of microlissencephaly. Exome sequencing identified seven different PNKP variants, including two novel ones. The c.1253_1269dup p.(Thr424GlyfsTer49) and c.1381_1383dup p.(Asn461dup) variants, each was recurrent in 10 patients (37%), while the c.1381_1383del p.(Asn461del) variant was recurrent in four patients (14.8%). Haplotype analysis confirmed that the p.Asn461dup variant has a founder effect in our population. No genotype-phenotype correlation was observed in our cohort. Our results provide 'microlissencephaly' as an emerging distinct phenotype linked to PNKP variants. As such, PNKP variants could be associated with four overlapping subgroups that lie along a unifying phenotypic continuum.
Biallelic MED29 variants cause pontocerebellar hypoplasia with cataracts.
Pontocerebellar hypoplasia (PCH) represents a group of disorders characterized by cerebellum and pons hypoplasia, variable cerebral involvement, microcephaly, severe global developmental delay (GDD), and seizures. We sought the genetic cause of PCH in two siblings. Genetic workup was performed by whole-exome sequencing followed by Sanger validation. Morpholino-knockdown zebrafish embryos with human wild-type gene rescue were used to assess cerebellar development and motor function. Transfected mouse hippocampal cultures and electroporated mouse embryos were employed to assess functional effects on neuronal morphology and development. Both patients presented with profound GDD, severe microcephaly, cataracts, and variably seizures. Their MRIs demonstrated marked cerebellar and pontine hypoplasia. Both were homozygous for a c.416T > C, p.(Leu139Pro) MED29 variant which was predicted to be pathogenic. Locomotion and cerebellar GABAergic neurons development were both impaired in MED29 Morpholino-knockdown zebrafish and rescued by human wild-type gene expression. ShRNA-knockdown of MED29 in mouse hippocampal neurons decreased neurite length and arborization in vitro, and caused defective embryonic neuronal migration in vivo. Overexpression of MED29 p.(Leu139Pro) was consistent with a loss-of-function. Taken together, the Mediator complex regulates transcription processes, and defects in particular subunits are associated with distinct neurodevelopmental phenotypes involving PCH. We conclude that MED29 is a novel risk gene for PCH.
The phenotyping dilemma in VRK1-related motor neuron disease: a Turkish family with young-onset amyotrophic lateral sclerosis caused by a novel mutation.
Objective: Vaccinia-related kinase 1 (VRK1)-related disease is an extremely rare autosomal recessive disorder primarily affecting the peripheral and/or central nervous system. In this report, we describe the genetic and clinical features of two siblings from a Turkish family presenting with an amyotrophic lateral sclerosis (ALS) phenotype due to a novel homozygous VRK1 mutation, and discuss the broad phenotypic spectrum associated with pathogenic variants in this gene. Methods: We analyzed the demographic data, clinical histories, neurological examinations, laboratory findings, and genetic results of 53 patients, including our cases, derived from 27 different reports. Results: Whole-exome sequencing identified a novel homozygous missense mutation, c.700A > G (p.Asn234Asp), in the VRK1 gene in two affected siblings. The characteristic features of the ALS phenotype included a recessive inheritance pattern, motor deficits with onset in the lower limbs, pyramidal tract signs, and a muscle magnetic resonance imaging (MRI) pattern demonstrating preferential involvement of the posterior compartments of the leg and thigh. The most common phenotypes associated with VRK1 mutations were ALS (18/53, 34%) and distal hereditary motor neuropathy (dHMN) (14/53, 26.4%), followed by pontocerebellar hypoplasia type 1 (7/53, 13.2%), hereditary motor and sensory neuropathy (5/53, 9.4%), autosomal recessive primary microcephaly with brain malformations (4/53, 7.5%), and spastic paraplegia (2/53, 3.8%). The ALS phenotype exhibited a significantly earlier mean age of onset compared to the dHMN phenotype (p = 0.015; 15.3 ± 11.5 and 27 ± 15.5 years, respectively). Conclusion: Our findings highlight the importance of investigating VRK1 mutations in patients with young-onset familial ALS. Furthermore, this report provides a systematic classification of the phenotype definitions associated with VRK1 mutations.
Expanding the genotypic and phenotypic spectrum of Egyptian children with maple syrup urine disease.
Maple Syrup Urine Disease (MSUD, OMIM# 248600) is an autosomal recessive inborn error of metabolism characterized by elevated branched chain amino acids (BCAA) leucine/isoleucine and valine in blood of affected children. The phenotypic and genotypic spectrum of MSUD is largely unreported in Egypt. We recruited ten patients (4 males/6 females, 2weeks-12years) from nine unrelated families with clinical and biochemical evidence of MSUD. We performed Sanger sequencing for the three most-commonly responsible genes: BCKDHA, BCKDHB and DBT and conducted exome sequencing for unresolved cases. Through Sanger sequencing, we detected eight homozygous pathogenic/likely pathogenic variants (four in BCKDHB, three in BCKDHA and one in DBT gene) in eight different families. The proband of family VI, who had no significant genetic findings by Sanger, had a peculiar phenotype and atypical radiological findings. His exome sequencing revealed a previously reported homozygous likely pathogenic variant in the RARS2 gene (NM_020320.5:c.1026G > A;p.(Met342Ile)) causing the mitochondrial-encephalopathy disorder pontocerebellar hypoplasia, type 6 (OMIM# 611523). Furthermore, the copy-number-variant analysis of the exome data revealed a biallelic duplication affecting exons 2-6 of the BCKDHB gene (GRCh38: Chr.6-g.80127496:80171441dup) evaluated as variant of uncertain significance but expected to cause a breakpoint and may disrupt gene function, which can explain the markedly elevated BCAA levels in the patient's blood. In conclusion, we expanded the genotypic and phenotypic spectrum of the disease and showed that aggressive intervention with specific treatment in the first few days of life resulted in normal development even in a developing country setting. Inclusion of MSUD in the national newborn screening program in Egypt is highly recommended.
Prenatal diagnosis of pontocerebellar hypoplasia with postnatal follow-up.
To describe the MR features enabling prenatal diagnosis of pontocerebellar hypoplasia (PCH). This was a retrospective single monocentre study. The inclusion criteria were decreased cerebellar biometry on dedicated neurosonography and available fetal Magnetic Resonance Imaging (MRI) with PCH diagnosis later confirmed either genetically or clinically on post-natal MRI or by autopsy. The exclusion criteria were non-available MRI and sonographic features suggestive of a known genetic or other pathologic diagnosis. The collected data were biometric or morphological imaging parameters, clinical outcome, termination of pregnancy (TOP), pathological findings and genetic analysis (karyotyping, chromosomal microarray, DNA sequencing targeted or exome). PCH was classified as classic, non-classic, chromosomal, or unknown type. Forty-two fetuses were diagnosed with PCH, of which 27 were referred for decreased transverse cerebellar diameter at screening ultrasound. Neurosonography and fetal MRI were performed at a mean gestational age of 29 + 4 and 31 + 0 weeks, respectively. Termination of pregnancy occurred. Pregnancy was terminated in 24 cases. Neuropathological examination confirmed the diagnosis in 24 cases and genetic testing identified abnormalities in 29 cases (28 families, 14 chromosomal anomaly). Classic PCH is associated with pontine atrophy and small MR measurements decreasing with advancing gestation. This is the first large series of prenatally diagnosed PCHs. Our study shows the essential contribution of fetal MRI to the prenatal diagnosis of PCH. Classic PCHs are particularly severe and are associated with certain MR features.
Publicações recentes
Pontocerebellar Hypoplasia Type 11 Case with a Novel Variant of TBC1D23 Gene: Case Report and Literature Review.
Diagnostic Clues and Pitfalls in Pontocerebellar Hypoplasia Type 2A.
46,XY differences of sex development in pontocerebellar hypoplasia type 7 (PCH7): two case reports and systematic review.
Genetic and clinical insights into pontocerebellar hypoplasia: Identification of novel variants in an Iranian cohort.
Connectome-seq: high-throughput mapping of neuronal connectivity at single-synapse resolution via barcode sequencing.
📚 EuropePMC251 artigos no totalmostrando 23
Refining the phenotypic spectrum of PNKP-related microcephaly: a study of 27 new patients.
Journal of medical geneticsBiallelic MED29 variants cause pontocerebellar hypoplasia with cataracts.
European journal of human genetics : EJHGThe phenotyping dilemma in VRK1-related motor neuron disease: a Turkish family with young-onset amyotrophic lateral sclerosis caused by a novel mutation.
Amyotrophic lateral sclerosis & frontotemporal degenerationExpanding the genotypic and phenotypic spectrum of Egyptian children with maple syrup urine disease.
Scientific reportsPrenatal diagnosis of pontocerebellar hypoplasia with postnatal follow-up.
Prenatal diagnosisClinical and genetic analysis of infants with pontocerebellar hypoplasia type 6 caused by RARS2 variations.
Epilepsia openNovel bi-allelic variants of CHMP1A contribute to pontocerebellar hypoplasia type 8: additional clinical and genetic evidence.
Frontiers in neurologyGenetic and prenatal diagnosis of a Chinese pedigree with pathogenic TOE1 variants causing pontocerebellar hypoplasia type 7.
The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal ObstetriciansReport of new variants in PPIL1 underlying type 14 pontocerebellar hypoplasia and their associated phenotypic manifestations in two fetuses.
American journal of medical genetics. Part ANovel compound heterozygous missense variants in TOE1 gene associated with pontocerebellar hypoplasia type 7.
GeneAnalysis of the Clinical Features and Imaging Findings of Pontocerebellar Hypoplasia Type 2D Caused by Mutations in SEPSECS Gene.
Cerebellum (London, England)A Rare Case of Pontocerebellar Hypoplasia Type 1B With Literature Review.
CureusA Neonate with a Diagnosis of Pontocerebellar Hypoplasia Type 6 Treated with Biotin and Developed Biotin Interference with Laboratory Thyroid Function Tests.
The American journal of case reportsTwo siblings with a novel variant of EXOSC3 extended phenotypic spectrum of pontocerebellar hypoplasia 1B to an exceptionally mild form.
BMJ case reportsPontocerebellar hypoplasia type 11: Does the genetic defect determine timing of cerebellar pathology?
European journal of medical geneticsLoss of Piccolo Function in Rats Induces Cerebellar Network Dysfunction and Pontocerebellar Hypoplasia Type 3-like Phenotypes.
The Journal of neuroscience : the official journal of the Society for Neuroscience[Clinical and genetic characteristics of 62 children with mitochondrial epilepsy].
Zhonghua er ke za zhi = Chinese journal of pediatricsTargeting ferroptosis: A novel therapeutic strategy for the treatment of mitochondrial disease-related epilepsy.
PloS onePontocerebellar hypoplasia type 1 for the neuropediatrician: Genotype-phenotype correlations and diagnostic guidelines based on new cases and overview of the literature.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism.
Brain : a journal of neurologyNovel homozygous RARS2 mutation in two siblings without pontocerebellar hypoplasia - further expansion of the phenotypic spectrum.
Orphanet journal of rare diseasesNeuropathologic Characterization of Pontocerebellar Hypoplasia Type 6 Associated With Cardiomyopathy and Hydrops Fetalis and Severe Multisystem Respiratory Chain Deficiency due to Novel RARS2 Mutations.
Journal of neuropathology and experimental neurologyTargeted carrier screening for four recessive disorders: high detection rate within a founder population.
European journal of medical geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Hipoplasia pontocerebelar tipo 14.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Hipoplasia pontocerebelar tipo 14
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Refining the phenotypic spectrum of PNKP-related microcephaly: a study of 27 new patients.
- Biallelic MED29 variants cause pontocerebellar hypoplasia with cataracts.
- The phenotyping dilemma in VRK1-related motor neuron disease: a Turkish family with young-onset amyotrophic lateral sclerosis caused by a novel mutation.
- Expanding the genotypic and phenotypic spectrum of Egyptian children with maple syrup urine disease.
- Prenatal diagnosis of pontocerebellar hypoplasia with postnatal follow-up.
- Pontocerebellar Hypoplasia Type 11 Case with a Novel Variant of TBC1D23 Gene: Case Report and Literature Review.
- Diagnostic Clues and Pitfalls in Pontocerebellar Hypoplasia Type 2A.
- 46,XY differences of sex development in pontocerebellar hypoplasia type 7 (PCH7): two case reports and systematic review.
- Genetic and clinical insights into pontocerebellar hypoplasia: Identification of novel variants in an Iranian cohort.
- Connectome-seq: high-throughput mapping of neuronal connectivity at single-synapse resolution via barcode sequencing.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:613274(Orphanet)
- OMIM OMIM:619301(OMIM)
- MONDO:0030258(MONDO)
- GARD:18032(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Hipoplasia pontocerebelar tipo 14
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)