Uma síndrome epiléptica que acontece na infância.
Introdução
O que você precisa saber de cara
Uma síndrome epiléptica que acontece na infância.
Tem tratamento?
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 120 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 281 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
37 genes identificados com associação a esta condição.
Curadoria gene-doença
fontes oficiaisPore-forming subunit of Nav1.3, a voltage-gated sodium (Nav) channel that directly mediates the depolarizing phase of action potentials in excitable membranes. Navs, also called VGSCs (voltage-gated sodium channels) or VDSCs (voltage-dependent sodium channels), operate by switching between closed and open conformations depending on the voltage difference across the membrane. In the open conformation they allow Na(+) ions to selectively pass through the pore, along their electrochemical gradient.
Cell membraneBasal cell membrane
Epilepsy, familial focal, with variable foci 4
An autosomal dominant form of epilepsy characterized by focal seizures arising from different cortical regions, including the temporal, frontal, parietal, and occipital lobes. Seizure types commonly include temporal lobe epilepsy, frontal lobe epilepsy, and nocturnal frontal lobe epilepsy. Some patients may have intellectual disability or autism spectrum disorders. Seizure onset usually occurs in the first or second decades, although later onset has been reported, and there is phenotypic variability within families. A subset of patients have structural brain abnormalities. Penetrance of the disorder is incomplete. FFEVF4 is characterized by onset of focal seizures in the first years of life.
Extracellular matrix serine protease secreted by pioneer neurons that plays a role in layering of neurons in the cerebral cortex and cerebellum by coordinating cell positioning during neurodevelopment. Regulates microtubule function in neurons and neuronal migration. Binding to the extracellular domains of lipoprotein receptors VLDLR and LRP8/APOER2 induces tyrosine phosphorylation of DAB1 and modulation of TAU phosphorylation. Affects migration of sympathetic preganglionic neurons in the spinal
Secreted, extracellular space, extracellular matrix
Lissencephaly 2
A classic type lissencephaly associated with ataxia, intellectual disability, seizures and abnormalities of the cerebellum, hippocampus and brainstem.
Monooxygenase that promotes depolymerization of F-actin by mediating oxidation of specific methionine residues on actin to form methionine-sulfoxide, resulting in actin filament disassembly and preventing repolymerization (PubMed:29343822). In the absence of actin, it also functions as a NADPH oxidase producing H(2)O(2) (PubMed:21864500, PubMed:26845023, PubMed:29343822). Acts as a cytoskeletal regulator that connects NEDD9 to intermediate filaments. Also acts as a negative regulator of apoptosi
CytoplasmCytoplasm, cytoskeletonEndosome membraneMidbody
Hormone regulating the release of corticotropin from pituitary gland (By similarity). Induces NLRP6 in intestinal epithelial cells, hence may influence gut microbiota profile (By similarity)
Secreted
Involved in normal synaptic function through regulation of Ca(2+) influx and neurotransmitter release in photoreceptor synaptic terminals and in auditory transmission. Modulator of CACNA1D and CACNA1F, suppressing the calcium-dependent inactivation and shifting the activation range to more hyperpolarized voltages (By similarity)
CytoplasmPresynapse
Cone-rod synaptic disorder, congenital non-progressive
A non-progressive retinal disorder characterized by stable low vision, nystagmus, photophobia, a normal or near-normal fundus appearance, and no night blindness.
Pore-forming subunit of Nav1.1, a voltage-gated sodium (Nav) channel that directly mediates the depolarizing phase of action potentials in excitable membranes. Navs, also called VGSCs (voltage-gated sodium channels) or VDSCs (voltage-dependent sodium channels), operate by switching between closed and open conformations depending on the voltage difference across the membrane. In the open conformation they allow Na(+) ions to selectively pass through the pore, along their electrochemical gradient.
Cell membrane
Generalized epilepsy with febrile seizures plus 2
A rare autosomal dominant, familial condition with incomplete penetrance and large intrafamilial variability. Patients display febrile seizures persisting sometimes beyond the age of 6 years and/or a variety of afebrile seizure types. This disease combines febrile seizures, generalized seizures often precipitated by fever at age 6 years or more, and partial seizures, with a variable degree of severity.
Component of the adaptor protein complex 2 (AP-2) (PubMed:12694563, PubMed:12952941, PubMed:14745134, PubMed:14985334, PubMed:15473838, PubMed:31104773). Adaptor protein complexes function in protein transport via transport vesicles in different membrane traffic pathways (PubMed:12694563, PubMed:12952941, PubMed:14745134, PubMed:14985334, PubMed:15473838, PubMed:31104773). Adaptor protein complexes are vesicle coat components and appear to be involved in cargo selection and vesicle formation (Pu
Cell membraneMembrane, coated pit
Intellectual developmental disorder, autosomal dominant 60, with seizures
An autosomal dominant disorder characterized by global developmental delay apparent in the first six months of life, followed by onset of seizures between 21 months and 4 years. Disease features include moderate-to-severe intellectual disability, poor speech, delayed walking, and ataxia.
Voltage-sensitive calcium channel that gives rise to T-type calcium currents. T-type calcium channels belong to the 'low-voltage activated (LVA)' group. A particularity of this type of channel is an opening at quite negative potentials, and a voltage-dependent inactivation (PubMed:27149520, PubMed:9670923, PubMed:9930755). T-type channels serve pacemaking functions in both central neurons and cardiac nodal cells and support calcium signaling in secretory cells and vascular smooth muscle (Probabl
Cell membrane
Epilepsy, idiopathic generalized 6
A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain.
May bind DNA
Nucleus
Alpha subunit of the heteropentameric ligand-gated chloride channel gated by Gamma-aminobutyric acid (GABA), a major inhibitory neurotransmitter in the brain (PubMed:23909897, PubMed:25489750, PubMed:29950725, PubMed:30602789). GABA-gated chloride channels, also named GABA(A) receptors (GABAAR), consist of five subunits arranged around a central pore and contain GABA active binding site(s) located at the alpha and beta subunit interface(s) (PubMed:29950725, PubMed:30602789). When activated by GA
Postsynaptic cell membraneCell membraneCytoplasmic vesicle membrane
Epilepsy, childhood absence 4
A subtype of idiopathic generalized epilepsy characterized by an onset at age 6-7 years, frequent absence seizures (several per day) and bilateral, synchronous, symmetric 3-Hz spike waves on EEG. Tonic-clonic seizures often develop in adolescence. Absence seizures may either remit or persist into adulthood.
Transcription factor involved in the control of neuronal proliferation and differentiation in the brain. Regulates dendrite development and branching, dendritic spine formation, and synaptogenesis in cortical layers II-III. Binds to DNA in a sequence-specific manner
Nucleus
Developmental and epileptic encephalopathy 67
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE67 is an autosomal dominant form characterized by onset of seizures in infancy. Later onset of seizures in childhood may occur in some patients.
Involved in neurite outgrowth by regulating cell-cell adhesion via the N-cadherin signaling pathway. May act by regulating expression of protein-coding genes, such as N-cadherins and integrin beta-1 (ITGB1)
NucleusCytoplasm
Intellectual developmental disorder, X-linked 98
A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. XLID98 patients show delayed psychomotor development, absent or poor speech development, and postnatal growth retardation, often with microcephaly. Some patients show autistic behavioral features, such as stereotypic hand movements and repetitive behaviors. Additional, more variable features include spasticity, axial hypotonia, seizures, drooling, gastroesophageal reflux, and lack of sphincter control.
Facilitative glucose transporter, which is responsible for constitutive or basal glucose uptake (PubMed:10227690, PubMed:10954735, PubMed:18245775, PubMed:19449892, PubMed:25982116, PubMed:27078104, PubMed:32860739). Has a very broad substrate specificity; can transport a wide range of aldoses including both pentoses and hexoses (PubMed:18245775, PubMed:19449892). Most important energy carrier of the brain: present at the blood-brain barrier and assures the energy-independent, facilitative trans
Cell membraneMelanosomePhotoreceptor inner segment
GLUT1 deficiency syndrome 1
A neurologic disorder showing wide phenotypic variability. The most severe 'classic' phenotype comprises infantile-onset epileptic encephalopathy associated with delayed development, acquired microcephaly, motor incoordination, and spasticity. Onset of seizures, usually characterized by apneic episodes, staring spells, and episodic eye movements, occurs within the first 4 months of life. Other paroxysmal findings include intermittent ataxia, confusion, lethargy, sleep disturbance, and headache. Varying degrees of cognitive impairment can occur, ranging from learning disabilities to severe intellectual disability.
Serine/threonine-protein kinase involved in various processes such as neuronal proliferation, differentiation, migration and programmed cell death. Extracellular stimuli such as pro-inflammatory cytokines or physical stress stimulate the stress-activated protein kinase/c-Jun N-terminal kinase (SAP/JNK) signaling pathway. In this cascade, two dual specificity kinases MAP2K4/MKK4 and MAP2K7/MKK7 phosphorylate and activate MAPK10/JNK3. In turn, MAPK10/JNK3 phosphorylates a number of transcription f
CytoplasmMembraneNucleusMitochondrion
Important modulator of glutamate signaling pathway
Cell membraneSynapse
Developmental and epileptic encephalopathy 37
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE37 is an autosomal recessive, severe form manifesting in the first years of life. Affected individuals show hyperkinetic movement disorder with choreoathetosis, spasticity, rigidity, intellectual disability, absent speech, and impaired volitional movements.
Major constituent of the PSD essential for postsynaptic signaling. Inhibitory regulator of the Ras-cAMP pathway. Member of the NMDAR signaling complex in excitatory synapses, it may play a role in NMDAR-dependent control of AMPAR potentiation, AMPAR membrane trafficking and synaptic plasticity. Regulates AMPAR-mediated miniature excitatory postsynaptic currents. Exhibits dual GTPase-activating specificity for Ras and Rap. May be involved in certain forms of brain injury, leading to long-term lea
Intellectual developmental disorder, autosomal dominant 5
A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD5 patients show global developmental delay with delayed motor development, hypotonia, moderate-to-severe intellectual disability, and severe language impairment. Epilepsy and autism can be present in some patients.
Receptor for adenosine (By similarity). The activity of this receptor is mediated by G proteins which activate adenylyl cyclase (By similarity)
Cell membrane
Voltage-sensitive calcium channels (VSCC) mediate the entry of calcium ions into excitable cells and are also involved in a variety of calcium-dependent processes, including muscle contraction, hormone or neurotransmitter release, gene expression, cell motility, cell division and cell death. The isoform alpha-1A gives rise to P and/or Q-type calcium currents. P/Q-type calcium channels belong to the 'high-voltage activated' (HVA) group and are specifically blocked by the spider omega-agatoxin-IVA
Cell membrane
Spinocerebellar ataxia 6
Spinocerebellar ataxia is a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA6 is an autosomal dominant cerebellar ataxia (ADCA), mainly caused by expansion of a CAG repeat in the coding region of CACNA1A. There seems to be a correlation between the repeat number and earlier onset of the disorder.
Acts as a ligand for the urokinase plasminogen activator surface receptor. Plays a role in angiogenesis by inducing endothelial cell migration and the formation of vascular network (cords). Involved in cellular migration and adhesion. Increases the phosphorylation levels of FAK. Interacts with and increases the mitogenic activity of HGF. Promotes synapse formation. May have a role in the perisylvian region, critical for language and cognitive development
SecretedCytoplasmCell surfaceSynapse
Rolandic epilepsy, impaired intellectual development, and speech dyspraxia, X-linked
A condition characterized by the association of rolandic seizures with oral and speech dyspraxia, and intellectual disability. Rolandic seizures occur during a period of significant brain maturation. During this time, dysfunction of neural network activities such as focal discharges may be associated with specific developmental disabilities resulting in specific cognitive impairments of language, visuo-spatial abilities or attention.
Endocrine hormone of the central and peripheral nervous systems that binds and activates the G protein-coupled receptors GALR1, GALR2, and GALR3. This small neuropeptide may regulate diverse physiologic functions including contraction of smooth muscle of the gastrointestinal and genitourinary tract, growth hormone and insulin release and adrenal secretion
Secreted
Epilepsy, familial temporal lobe, 8
A focal form of epilepsy characterized by recurrent seizures that arise from foci within the temporal lobe. Seizures are usually accompanied by sensory symptoms, most often auditory in nature.
Component of neuronal acetylcholine receptors (nAChRs) that function as pentameric, ligand-gated cation channels with high calcium permeability among other activities. nAChRs are excitatory neurotrasnmitter receptors formed by a collection of nAChR subunits known to mediate synaptic transmission in the nervous system and the neuromuscular junction. Each nAchR subunit confers differential attributes to channel properties, including activation, deactivation and desensitization kinetics, pH sensiti
Synaptic cell membraneCell membrane
Epilepsy, nocturnal frontal lobe, 3
An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements.
Component of neuronal acetylcholine receptors (nAChRs) that function as pentameric, ligand-gated cation channels with high calcium permeability among other activities. nAChRs are excitatory neurotrasnmitter receptors formed by a collection of nAChR subunits known to mediate synaptic transmission in the nervous system and the neuromuscular junction. Each nAchR subunit confers differential attributes to channel properties, including activation, deactivation and desensitization kinetics, pH sensiti
Synaptic cell membraneCell membrane
Epilepsy, nocturnal frontal lobe, 1
An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements.
Sodium-activated K(+) channel (PubMed:37494189). Acts as an important mediator of neuronal membrane excitability (PubMed:37494189). Contributes to the delayed outward currents (By similarity). Regulates neuronal bursting in sensory neurons (By similarity). Contributes to synaptic development and plasticity (By similarity)
Cell membrane
Developmental and epileptic encephalopathy 14
A rare epileptic encephalopathy of infancy that combines pharmacoresistant seizures with developmental delay. This severe neurologic disorder is characterized by onset in the first 6 months of life of refractory focal seizures and arrest of psychomotor development. Ictal EEG shows discharges that arise randomly from various areas of both hemispheres and migrate from one brain region to another.
ATP-dependent chromatin-remodeling factor that specifically binds to the promoter of target genes, leading to chromatin remodeling, possibly by promoting deposition of histone H3.3. Involved in myogenesis via interaction with MYOD1: binds to myogenic gene regulatory sequences and mediates incorporation of histone H3.3 prior to the onset of myogenic gene expression, promoting their expression (By similarity)
Nucleus
Developmental and epileptic encephalopathy 94
A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE94 is an autosomal dominant, severe form characterized by onset of multiple seizure types in the first few years of life.
Regulates voltage-gated potassium channels assembled from KCNA1, KCNA4 and KCNAB1. It slows down channel inactivation by precluding channel closure mediated by the KCNAB1 subunit. Ligand for ADAM22 that positively regulates synaptic transmission mediated by AMPA-type glutamate receptors (By similarity). Plays a role in suppressing the production of MMP1/3 through the phosphatidylinositol 3-kinase/ERK pathway. May play a role in the control of neuroblastoma cell survival
SecretedSynapseCytoplasmGolgi apparatusEndoplasmic reticulum
Epilepsy, familial temporal lobe, 1
A focal form of epilepsy characterized by recurrent seizures that arise from foci within the temporal lobe. Seizures are usually accompanied by sensory symptoms, most often auditory in nature.
As a component of the GATOR1 complex functions as an inhibitor of the amino acid-sensing branch of the mTORC1 pathway (PubMed:23723238, PubMed:29590090, PubMed:35338845). In response to amino acid depletion, the GATOR1 complex has GTPase activating protein (GAP) activity and strongly increases GTP hydrolysis by RagA/RRAGA (or RagB/RRAGB) within heterodimeric Rag complexes, thereby turning them into their inactive GDP-bound form, releasing mTORC1 from lysosomal surface and inhibiting mTORC1 signa
Lysosome membrane
Catalyzes the hydrolysis of GTP and utilizes this energy to mediate vesicle scission and participates in many forms of endocytosis, such as clathrin-mediated endocytosis or synaptic vesicle endocytosis as well as rapid endocytosis (RE) (PubMed:15703209, PubMed:20428113, PubMed:29668686, PubMed:8101525, PubMed:8910402, PubMed:9362482). Associates to the membrane, through lipid binding, and self-assembles into rings and stacks of interconnected rings through oligomerization to form a helical polym
Cell membraneMembrane, clathrin-coated pitCytoplasmic vesiclePresynapseCytoplasmic vesicle, secretory vesicle, chromaffin granule
Developmental and epileptic encephalopathy 31A
An autosomal dominant epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent.
Catalytic component of the GATOR1 complex, a multiprotein complex that functions as an inhibitor of the amino acid-sensing branch of the mTORC1 pathway (PubMed:23723238, PubMed:29590090, PubMed:35338845, PubMed:38006878). In response to amino acid depletion, the GATOR1 complex has GTPase activating protein (GAP) activity and strongly increases GTP hydrolysis by RagA/RRAGA (or RagB/RRAGB) within heterodimeric Rag complexes, thereby turning them into their inactive GDP-bound form, releasing mTORC1
Lysosome membrane
May be involved in the proteolytic inactivation of enkephalins and neurotensin in some brain areas. May convert inactive angiotensin I into the biologically active angiotensin II (PubMed:18178555). Releases a C-terminal amino acid, with preference for large hydrophobic C-terminal amino acids and shows only very weak activity toward small amino acids and histidine (PubMed:20855895)
Secreted, extracellular space, extracellular matrix
Mediates transport of gamma-aminobutyric acid (GABA) together with sodium and chloride and is responsible for the reuptake of GABA from the synapse (PubMed:30132828). The translocation of GABA, however, may also occur in the reverse direction leading to the release of GABA (By similarity). The direction and magnitude of GABA transport is a consequence of the prevailing thermodynamic conditions, determined by membrane potential and the intracellular and extracellular concentrations of Na(+), Cl(-
Cell membranePresynapse
Myoclonic-atonic epilepsy
A form of epilepsy characterized by myoclonic-atonic and absence seizures, appearing in early childhood. Patients have delayed development before the onset of seizures and show varying degrees of intellectual disability following seizure onset.
As a component of the GATOR1 complex functions as an inhibitor of the amino acid-sensing branch of the mTORC1 pathway (PubMed:23723238, PubMed:25457612, PubMed:29590090, PubMed:29769719, PubMed:31548394, PubMed:35338845). In response to amino acid depletion, the GATOR1 complex has GTPase activating protein (GAP) activity and strongly increases GTP hydrolysis by RagA/RRAGA (or RagB/RRAGB) within heterodimeric Rag complexes, thereby turning them into their inactive GDP-bound form, releasing mTORC1
Lysosome membraneCytoplasm, cytosolCytoplasm, perinuclear region
Epilepsy, familial focal, with variable foci 1
An autosomal dominant form of epilepsy characterized by focal seizures arising from different cortical regions in different family members. Many patients have an aura and show automatisms during the seizures, whereas others may have nocturnal seizures. There is often secondary generalization. Some patients show abnormal interictal EEG, and some patients may have intellectual disability or autism spectrum disorders. Seizure onset usually occurs in the first or second decades, although later onset has been reported, and there is phenotypic variability within families. Penetrance of the disorder is incomplete.
Beta subunit of the heteropentameric ligand-gated chloride channel gated by gamma-aminobutyric acid (GABA), a major inhibitory neurotransmitter in the brain (PubMed:14993607, PubMed:18514161, PubMed:22243422, PubMed:22303015, PubMed:24909990, PubMed:26950270, PubMed:30602789). GABA-gated chloride channels, also named GABA(A) receptors (GABAAR), consist of five subunits arranged around a central pore and contain GABA active binding site(s) located at the alpha and beta subunit interface(s) (PubMe
Postsynaptic cell membraneCell membraneCytoplasmic vesicle membrane
Epilepsy, childhood absence 5
A subtype of idiopathic generalized epilepsy characterized by an onset at age 6-7 years, frequent absence seizures (several per day) and bilateral, synchronous, symmetric 3-Hz spike waves on EEG. Tonic-clonic seizures often develop in adolescence. Absence seizures may either remit or persist into adulthood.
Potassium channel activated by both membrane depolarization or increase in cytosolic Ca(2+) that mediates export of K(+) (PubMed:14523450, PubMed:29330545, PubMed:31152168). It is also activated by the concentration of cytosolic Mg(2+). Its activation dampens the excitatory events that elevate the cytosolic Ca(2+) concentration and/or depolarize the cell membrane. It therefore contributes to repolarization of the membrane potential. Plays a key role in controlling excitability in a number of sys
Cell membrane
Paroxysmal non-kinesigenic dyskinesia 3 with or without generalized epilepsy
An autosomal dominant neurologic disorder characterized by absence seizures, generalized tonic-clonic seizures, paroxysmal nonkinesigenic dyskinesia and involuntary dystonic or choreiform movements. Onset is usually in childhood. Patients may have seizures only, dyskinesia only, or both.
May act as a GTPase-activating protein for Rab family protein(s) (PubMed:20727515, PubMed:20797691). Involved in neuronal projections development, probably through a negative modulation of ARF6 function (PubMed:20727515). Involved in the regulation of synaptic vesicle trafficking (PubMed:31257402)
Cell membraneCytoplasmCytoplasmic vesicle membranePresynapse
Familial infantile myoclonic epilepsy
A subtype of idiopathic epilepsy starting in early infancy and manifesting as myoclonic seizures, febrile convulsions, and tonic-clonic seizures.
Component of neuronal acetylcholine receptors (nAChRs) that function as pentameric, ligand-gated cation channels with high calcium permeability among other activities. nAChRs are excitatory neurotrasnmitter receptors formed by a collection of nAChR subunits known to mediate synaptic transmission in the nervous system and the neuromuscular junction. Each nAchR subunit confers differential attributes to channel properties, including activation, deactivation and desensitization kinetics, pH sensiti
Synaptic cell membraneCell membrane
Epilepsy, nocturnal frontal lobe, 4
An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking.
Component of N-methyl-D-aspartate (NMDA) receptors (NMDARs) that function as heterotetrameric, ligand-gated cation channels with high calcium permeability and voltage-dependent block by Mg(2+) (PubMed:20890276, PubMed:23933818, PubMed:23933819, PubMed:23933820, PubMed:24504326, PubMed:26875626, PubMed:26919761, PubMed:28242877, PubMed:36117210, PubMed:38538865, PubMed:8768735). NMDARs participate in synaptic plasticity for learning and memory formation by contributing to the slow phase of excita
Cell projection, dendritic spineCell membraneSynapsePostsynaptic cell membraneCytoplasmic vesicle membrane
Epilepsy, focal, with speech disorder and with or without impaired intellectual development
An autosomal dominant, highly variable neurologic disorder. Features range from severe early-onset seizures associated with delayed psychomotor development, persistent speech difficulties, and intellectual disability to a more benign entity characterized by childhood onset of mild or asymptomatic seizures associated with transient speech difficulties followed by remission of seizures in adolescence and normal psychomotor development. The disorder encompasses several clinical entities, including Landau-Kleffner syndrome, epileptic encephalopathy with continuous spike and wave during slow-wave sleep, autosomal dominant rolandic epilepsy, intellectual disability and speech dyspraxia, and benign epilepsy with centrotemporal spikes.
Gamma subunit of the heteropentameric ligand-gated chloride channel gated by gamma-aminobutyric acid (GABA), a major inhibitory neurotransmitter in the brain (PubMed:14993607, PubMed:16412217, PubMed:23909897, PubMed:2538761, PubMed:25489750, PubMed:27864268, PubMed:29950725, PubMed:30602789). GABA-gated chloride channels, also named GABA(A) receptors (GABAAR), consist of five subunits arranged around a central pore and contain GABA active binding site(s) located at the alpha and beta subunit in
Postsynaptic cell membraneCell membraneCell projection, dendriteCytoplasmic vesicle membrane
Developmental and epileptic encephalopathy 74
A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE74 is an autosomal dominant form with onset in the first year of life.
Medicamentos e terapias
Mecanismo: Sodium channel alpha subunit blocker
Mecanismo: Cannabinoid CB1 receptor negative allosteric modulator
Mecanismo: Sodium channel alpha subunit blocker
Variantes genéticas (ClinVar)
910 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 2 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
67 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome epilepsia de início na infância
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Intellectual function and psychiatric comorbidities in patients with epilepsy with eyelid myoclonia.
Epilepsy with Eyelid Myoclonia (EEM) is a rare childhood-onset epilepsy syndrome. There is limited data about cognitive function and psychiatric comorbidities in patients with EEM. A database of 134 patients with EEM was reviewed for patients who underwent neuropsychological testing. Psychiatric comorbidities and psychometric test scores were identified. Group comparison was performed between those who underwent neuropsychological testing and those who did not. In addition, we evaluated whether clinical factors were associated with IQ score. Fourteen patients underwent neuropsychological testing (12 females, 85.7%), with a median age at testing of 17 (range 7-22). Median IQ was 79 (range 56-110); 7 patients had below average IQ. Other median results of neuropsychometric measures were: Verbal Comprehension Index 85.5 (range 66-116), Perceptual Reasoning Index or Visual Spatial Index 81.5 (range 67-100), Working Memory Index 77 (range 54-100), Processing Speed Index 84 (range 53-94), and Reading Standardized Scores 84 (range 64-126). Common psychiatric comorbidities were anxiety (n = 10), depression (n = 7), ADHD (n = 6), and autism (n = 2). Those who underwent neuropsychological testing had a younger age of epilepsy onset, longer follow-up at our institution, and were more likely to have myoclonic seizures or psychosis than those who did not undergo neuropsychological testing. No clinical factors were statistically associated with IQ score. EEM is associated with a wide range of cognitive abilities, with half of our patients having a below average IQ. Psychiatric comorbidities were common. Identifying cognitive impairment and psychiatric comorbidities is crucial to implement appropriate management strategies.
Current and Future Treatment Strategies in Developmental and/or Epileptic Encephalopathy With Spike-Wave Activation in Sleep (DEE-SWAS): A Time for Precision Medicine?
Developmental and/or epileptic encephalopathy with spike-wave activation in sleep (D/EE-SWAS) is a childhood-onset epilepsy syndrome characterized by cognitive regression or stagnation and marked activation of epileptiform activity during sleep. DEE-SWAS refers to cases with pre-existing neurodevelopmental disorders, whereas EE-SWAS is applied when development was initially normal before the onset of epileptic encephalopathy. This syndrome comprises approximately 0.2%-1.3% of all pediatric epilepsies. D/EE-SWAS etiology includes structural anomalies and autoimmune and genetic causes, although etiology frequently remains unknown. The active epileptic process in a developing brain results in impairment of cognitive functions and behavior. For this reason, early recognition of the electroclinical syndrome and treatment initiation is extremely relevant for the long-term cognitive outcome. Typically, the available therapeutic strategies consisted of low-quality evidence and limited effectiveness, such as combinations of antiseizure medications and steroids, which were based on syndromic diagnoses rather than etiology-driven hypotheses. Over the last years, treatment has been shifting toward precision medicine approaches, with an increasing proportion of genetic diagnosis, new evidence supporting the efficacy of the surgical option in selected patients, and specific targeted treatments, such as l-serine in GRIN-related disorders. Additionally, this coexists with ongoing clinical trials with syndrome-specific design for D/EE-SWAS. This narrative review aims to summarize the evidence on treatments for D/EE-SWAS, provide updates on drugs currently in development, and explore precision medicine approaches for this syndrome, seeking to combine both syndrome- and etiology-driven treatment strategies.
Epilepsy with myoclonic-atonic seizures: an update on genetic causes, nosological limits, and treatment strategies.
Epilepsy with myoclonic-atonic seizures is a childhood-onset epilepsy syndrome characterised by a range of seizure types, including myoclonic-atonic, atonic, myoclonic, absence, and generalised tonic-clonic seizures. The causes and outcomes of this syndrome are highly variable, with many uncertainties surrounding its classification and prognosis. Traditional antiseizure medications and the ketogenic diet remain the main treatment options. Although two-thirds of children attain remission from seizures without cognitive or behavioural sequelae, some continue to have drug-resistant seizures, intellectual disability, and behavioural problems. The identification of single-gene causes in a substantial subset of patients highlights the importance of genetic testing for development of personalised treatment strategies. However, diagnostic complexities have hindered the development of trials for new therapies. Better recognition of the distinct features of epilepsy with myoclonic-atonic seizures, combined with advances in molecular genetic testing, will pave the way for more focused clinical research and drug development. Future studies should aim to identify genetic causes and tailor treatment options, offering hope for improved long-term outcomes.
Efficacy and Tolerability of Lacosamide in Lennox-Gastaut Syndrome: A Systematic Review and Meta-analysis.
Purpose Lennox-Gastaut syndrome (LGS) is one of the most difficult to treat childhood-onset epileptic encephalopathies. There is growing evidence that lacosamide is safe and efficacious in patients and adults with refractory epilepsy. However, the evidence regarding the efficacy of lacosamide in LGS is controversial so far. We aimed to evaluate the efficacy and tolerability of lacosamide in patients with LGS. Methods We conducted a systematic review on MEDLINE, EMBASE, COCHRANE CENTRAL, Google Scholar, and Web of Science, collating all available literature till July 31, 2020. The qualitative review included case reports, case series, and both controlled/uncontrolled trials as well as retrospective studies, but for determining pooled estimates, we only included studies with a sample size of 5 or more. The primary outcome was the efficacy of lacosamide in patients with LGS. Clinical variables related to efficacy and adverse events attributed to lacosamide were extracted from each publication. The pooled estimate of variables related to these parameters was performed using a random-effect model. Results Of the 68 items identified by the search, 14 were reviewed as full-text. Eleven articles including two prospective and six retrospective studies fulfilled eligibility criteria and described outcomes in 81 patients (42 adults, 39 children, 60% male, range-1.4-61 years). On average, 35.2%, 27.9%, 7.3%, and 29.4% patients had > 50% reduction, < 50% reduction, no change, and worsening of seizure frequency, respectively. Although 36% of patients had adverse events like somnolence, behavioral abnormalities including irritability, aggressiveness, nausea, tremor, memory problems, dizziness, gastrointestinal discomfort, vomiting, and weight loss, no serious adverse events were noted. Conclusion The evidence available in the current literature is not sufficient to support or refute the use of lacosamide in patients with LGS. Although it is one of the possible therapeutic options worth exploring in patients with LGS, caution is still necessary, as there are reports of worsening of seizure frequency in some patients.
PCDH19-Related Epilepsy Syndrome: A Comprehensive Clinical Review.
PCDH19-related epilepsy is a distinct childhood-onset epilepsy syndrome characterized by brief clusters of febrile and afebrile seizures with onset primarily before the age of three years, cognitive impairment, autistic traits, and behavioral abnormalities. PCDH19 gene is located in Xq22 and produces nonclustered delta protocadherin. This disorder primarily manifests in heterozygote females due to random X chromosome inactivation leading to somatic mosaicism and abnormal cellular interference between cells with and without delta-protocadherin. This article reviews the clinical features based on a comprehensive literature review (MEDLINE using PubMed and OvidSP vendors with appropriate keywords to incorporate recent evidence), personal practice, and experience. Significant progress has been made in the past 10 years, including identification of the gene responsible for the condition, characterization of clinical phenotypes, and development of animal models. More rigorous studies involving quality-of-life measures as well as standardized neuropsychiatric testing are necessary to understand the full spectrum of the disease. The recent discovery of allopregnanolone deficiency in patients with PCDH19-related epilepsy leads to opportunities in precision therapy. A phase 3 clinical study is currently active to evaluate the efficacy, safety, and tolerability of adjunctive ganaxolone (an allopregnanolone analog) therapy.
Publicações recentes
Intellectual function and psychiatric comorbidities in patients with epilepsy with eyelid myoclonia.
Current and Future Treatment Strategies in Developmental and/or Epileptic Encephalopathy With Spike-Wave Activation in Sleep (DEE-SWAS): A Time for Precision Medicine?
Epilepsy with myoclonic-atonic seizures: an update on genetic causes, nosological limits, and treatment strategies.
Efficacy and Tolerability of Lacosamide in Lennox-Gastaut Syndrome: A Systematic Review and Meta-analysis.
PCDH19-Related Epilepsy Syndrome: A Comprehensive Clinical Review.
📚 EuropePMCmostrando 7
Intellectual function and psychiatric comorbidities in patients with epilepsy with eyelid myoclonia.
Epilepsy & behavior : E&BCurrent and Future Treatment Strategies in Developmental and/or Epileptic Encephalopathy With Spike-Wave Activation in Sleep (DEE-SWAS): A Time for Precision Medicine?
Pediatric neurologyEpilepsy with myoclonic-atonic seizures: an update on genetic causes, nosological limits, and treatment strategies.
The Lancet. NeurologyEfficacy and Tolerability of Lacosamide in Lennox-Gastaut Syndrome: A Systematic Review and Meta-analysis.
Journal of neurosciences in rural practicePCDH19-Related Epilepsy Syndrome: A Comprehensive Clinical Review.
Pediatric neurologyStiripentol for the treatment of seizures in Dravet syndrome.
Expert review of clinical pharmacologyVagus nerve stimulation for genetic epilepsy with febrile seizures plus (GEFS+) accompanying seizures with impaired consciousness.
Epilepsy & behavior case reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome epilepsia de início na infância.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome epilepsia de início na infância
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Intellectual function and psychiatric comorbidities in patients with epilepsy with eyelid myoclonia.
- Current and Future Treatment Strategies in Developmental and/or Epileptic Encephalopathy With Spike-Wave Activation in Sleep (DEE-SWAS): A Time for Precision Medicine?
- Epilepsy with myoclonic-atonic seizures: an update on genetic causes, nosological limits, and treatment strategies.
- Efficacy and Tolerability of Lacosamide in Lennox-Gastaut Syndrome: A Systematic Review and Meta-analysis.
- PCDH19-Related Epilepsy Syndrome: A Comprehensive Clinical Review.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:98259(Orphanet)
- MONDO:0020072(MONDO)
- Epilepsia(PCDT · Ministério da Saúde)
- GARD:19437(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q5382985(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
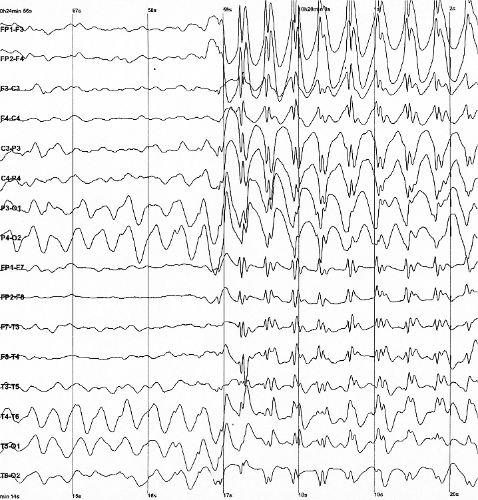
Síndrome epilepsia de início na infância
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos (literatura)
- fonte: Orphanet
- Reposicionamento
- fonte: Drug Repurposing Hub