A cistinose nefróptica juvenil é a forma intermediária da cistinose, tanto em relação à gravidade quanto à idade em que a doença se manifesta. É uma doença metabólica caracterizada pelo acúmulo de uma substância chamada cistina dentro de pequenos compartimentos nas células, conhecidos como lisossomos. Esse acúmulo causa danos em diversos órgãos e tecidos, especialmente nos rins e nos olhos.
Introdução
O que você precisa saber de cara
A cistinose nefróptica juvenil é a forma intermediária da cistinose, tanto em relação à gravidade quanto à idade em que a doença se manifesta. É uma doença metabólica caracterizada pelo acúmulo de uma substância chamada cistina dentro de pequenos compartimentos nas células, conhecidos como lisossomos. Esse acúmulo causa danos em diversos órgãos e tecidos, especialmente nos rins e nos olhos.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 20 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 46 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisCystine/H(+) symporter that mediates export of cystine, the oxidized dimer of cysteine, from lysosomes (PubMed:11689434, PubMed:15128704, PubMed:18337546, PubMed:22232659, PubMed:29467429, PubMed:33208952, PubMed:36113465). Plays an important role in melanin synthesis by catalyzing cystine export from melanosomes, possibly by inhibiting pheomelanin synthesis (PubMed:22649030). In addition to cystine export, also acts as a positive regulator of mTORC1 signaling in kidney proximal tubular cells, v
Lysosome membraneMelanosome membraneCell membrane
Cystinosis, nephropathic type
A form of cystinosis, a lysosomal storage disease due to defective transport of cystine across the lysosomal membrane. This results in cystine accumulation and crystallization in the cells causing widespread tissue damage. The classical nephropathic form has onset in the first year of life and is characterized by a polyuro-polydipsic syndrome, marked height-weight growth delay, generalized impaired proximal tubular reabsorptive capacity, with severe fluid-electrolyte balance alterations, renal failure, ocular symptoms and other systemic complications.
Variantes genéticas (ClinVar)
702 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 605 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Cistinose nefropática juvenil
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Multinucleated podocytes as a clue to diagnosis of juvenile nephropathic cystinosis.
BACKGROUND : Cystinosis is a rare autosomal recessive lysosomal disorder that mainly affects the kidney and eye. Early treatment with cysteamine significantly improves the prognosis. However, early diagnosis of cystinosis, especially the juvenile nephropathic form, remains challenging because typical symptoms only become apparent in adulthood. We herein describe a 13-year-old girl who presented with proteinuria only but was diagnosed with juvenile nephropathic cystinosis based on multinucleated podocytes in her kidney biopsy specimen. We also studied the nephropathology of another case to determine the features of the multinucleated podocytes. CASE DIAGNOSIS: A previously healthy 13-year-old girl presented to our hospital because proteinuria had been detected in her school urine screening. She had been noted to have proteinuria on her school urine screening when she was 11 years of age but there was no consultation with her physician at that time. She was asymptomatic and had no other abnormalities on examination other than a relatively high urinary β-2 microglobulin level. Her kidney biopsy showed 15 multinucleated podocytes in 34 glomeruli, and the mean number of nuclei per multinucleated podocyte was 4.4. Ophthalmological examination showed cystine crystals in her cornea. Her white blood cell cystine level was high, and she was diagnosed with juvenile nephropathic cystinosis. She started oral cysteamine treatment and showed almost no progression of the disease after 2 years. In another patient with juvenile nephropathic cystinosis, there were 25 multinucleated podocytes in 63 glomeruli, and the mean number of nuclei per multinucleated podocyte was 2.9. CONCLUSION: Early diagnosis is crucial to improve the prognosis of patients with cystinosis. This report emphasizes the importance of recognizing the unique pathological feature of multinucleated podocytes as an essential clue to the diagnosis of cystinosis.
The Pitfall of White Blood Cell Cystine Measurement to Diagnose Juvenile Cystinosis.
Cystinosis is an autosomal recessive lysosomal storage disease, caused by mutations in the CTNS gene, resulting in multi-organ cystine accumulation. Three forms of cystinosis are distinguished: infantile and juvenile nephropathic cystinosis affecting kidneys and other organs such as the eyes, endocrine system, muscles, and brain, and adult ocular cystinosis affecting only the eyes. Currently, elevated white blood cell (WBC) cystine content is the gold standard for the diagnosis of cystinosis. We present a patient with proteinuria at adolescent age and corneal cystine crystals, but only slightly elevated WBC cystine levels (1.31 ½ cystine/mg protein), precluding the diagnosis of nephropathic cystinosis. We demonstrate increased levels of cystine in skin fibroblasts and urine-derived kidney cells (proximal tubular epithelial cells and podocytes), that were higher than the values observed in the WBC and healthy control. CTNS gene analysis shows the presence of a homozygous missense mutation (c.590 A > G; p.Asn177Ser), previously described in the Arab population. Our observation underlines that low WBC cystine levels can be observed in patients with juvenile cystinosis, which may delay the diagnosis and timely administration of cysteamine. In such patients, the diagnosis can be confirmed by cystine measurement in slow-dividing cells and by molecular analysis of the CTNS gene.
More than tubular dysfunction: cystinosis and kidney outcomes.
Cystinosis is a lysosomal storage disease that affects many tissues. Its prognosis depends predominantly on kidney involvement. Cystinosis has three clinical forms: nephropathic infantile, nephropathic juvenile and non-nephropathic adult. Proximal tubular dysfunction is prominent in the infantile form, whereas a combination of glomerular and tubular alterations are observed in the juvenile form. Thirty-six children with nephropathic cystinosis were included in the study. Clinical features, molecular genetic diagnoses, and kidney outcomes of the patients were evaluated. Twenty-one children (58.3%) were male. The median age at diagnosis was 18.5 months. Twenty-eight patients (77.8%) had infantile nephropathic cystinosis, while eight (22.2%) had juvenile nephropathic cystinosis. An acute rapid deterioration of the kidney function with proteinuria, hypoalbuminemia, and nephrotic syndrome, was observed in 37.5% of patients with the juvenile form. The mean estimated glomerular filtration rate (eGFR) was 82.31 ± 37.45 ml/min/1.73m2 at diagnosis and 63.10 ± 54.60 ml/min/1.73m2 at the last visit (p = 0.01). Six patients (16.6%) had kidney replacement therapy (KRT) at the last visit. The median age of patients with kidney failure was 122 months. Patients with a spot urine protein/creatinine ratio < 6 mg/mg at the time of diagnosis had better kidney outcomes (p = 0.01). The most common allele was c.451A>G (32.6%). The patients with the most common mutation tended to have higher mean eGFR and lower leukocyte cystine levels than patients with other mutations. Glomerulonephritis may be a frequent finding in addition to the well-known tubular dysfunction in patients with cystinosis. Furthermore, our results highlight that the presence of severe proteinuria at the time of diagnosis is a relevant prognostic factor for kidney survival.
Testicular function in males with infantile nephropathic cystinosis.
Do males with the rare lysosomal storage disease infantile nephropathic cystinosis (INC) have a chance of biological fatherhood? Cryostorage of semen could be an option for approximately 20% of young males with INC, with surgical sperm retrieval from the centre of the testes providing additional opportunities for fatherhood. Biallelic mutations in the cystinosin (CTNS) gene in INC cause dysfunction in cystine transport across lysosomal membranes and cystine accumulation throughout the body. Spontaneous paternity in cystinosis has not been described, despite the availability of cysteamine treatment. Azoospermia has been diagnosed in small case series of males with INC. ART using ICSI requires few spermatozoa, either from semen or extracted surgically from the testes of azoospermic men. However, there is limited evidence to suggest this could be successful in INC. In this prospective cohort study performed between 2018 and 2019, we performed a cross-sectional investigation of 18 male patients with INC to delineate endocrine and spermatogenic testicular function. Serum hormone levels, semen samples (according to World Health Organization 2010 standards), and testicular ultrasound images were analysed in 18 male patients aged 15.4-40.5 years. Surgical sperm extraction was performed in two, and their testicular biopsies were investigated by light and electron microscopy. Past adherence to cysteamine treatment was assessed from medical record information, using a composite scoring system. Adherence to cysteamine treatment was high in most patients. Testicular volumes and testosterone levels were in the normal ranges, with the exception of two and three older patients, respectively. Serum LH levels were above the normal range in all subjects aged ≥20 years. FSH levels were elevated in all but four males: three with spermatozoa in semen and one adolescent. Inhibin B levels were shown to be lower in older men. Testicular ultrasound revealed signs of obstruction in 67% of patients. Reduced fructose and zinc seminal markers were found in 33%, including two patients with azoospermia who underwent successful surgical sperm retrieval. Histology identified fully preserved spermatogenesis in the centre of their testes, but also tubular atrophy and lysosomal overload in Sertoli and Leydig cells of the testicular periphery. Limitations of this study are the small number of assessed patients and the heterogeneity of their dysfunction in cystine transport across lysosomal membranes. This study suggests that testicular degeneration in cystinosis results from the lysosomal overload of Sertoli and Leydig cells of the testicular periphery, and that this can possibly be delayed, but not prevented, by good adherence to cysteamine treatment. Endocrine testicular function in INC may remain compensated until the fourth decade of life; however, azoospermia may occur during adolescence. Cryostorage of semen could be an option for approximately 20% of young males with INC, with surgical sperm retrieval providing additional opportunities for biological fatherhood. This work was supported by the Cystinosis Foundation Germany. The authors have no competing interests to declare. n/a.
Nephropathic cystinosis presenting with uveitis: Report of a "Can't See, Can't Pee" situation.
Nephropathic cystinosis is a rare autosomal recessive lysosomal disease characterized by accumulation of pathognomonic cystine crystals in renal and other tissues of the body. Cystinosis is caused by mutant cystinosin, the cystine transport protein located in lysosomal membranes, leading to systemic deposits of cystine and resultant end organ damage. Cystinosis is rarer in Asians than Caucasians with only a handful of cases reported from India to date. Due to its extreme rarity and clinically insidious presentation in contrast to the infantile form, the diagnosis of juvenile nephropathic cystinosis is frequently delayed or overlooked. Moreover, routine processing and sectioning of paraffin embedded tissues dissolves cystine crystals, making it difficult to diagnose this condition on light microscopic examination alone, mandating electron microscopic (EM) analysis of renal biopsies for an accurate diagnosis of this condition. We describe a case of juvenile nephropathic cystinosis presenting with uveitis and photophobia in a 17-year-old Indian male, diagnosed after EM examination of the patient's renal biopsy for evaluation of nephrotic syndrome. While highlighting the diagnostic utility of EM, we describe a few histopathologic clues which can prompt inclusion of EM analysis of renal biopsies in this setting.
Publicações recentes
Multinucleated podocytes as a clue to diagnosis of juvenile nephropathic cystinosis.
The Pitfall of White Blood Cell Cystine Measurement to Diagnose Juvenile Cystinosis.
More than tubular dysfunction: cystinosis and kidney outcomes.
Testicular function in males with infantile nephropathic cystinosis.
Nephropathic cystinosis presenting with uveitis: Report of a "Can't See, Can't Pee" situation.
📚 EuropePMC3 artigos no totalmostrando 7
Multinucleated podocytes as a clue to diagnosis of juvenile nephropathic cystinosis.
Pediatric nephrology (Berlin, Germany)The Pitfall of White Blood Cell Cystine Measurement to Diagnose Juvenile Cystinosis.
International journal of molecular sciencesMore than tubular dysfunction: cystinosis and kidney outcomes.
Journal of nephrologyTesticular function in males with infantile nephropathic cystinosis.
Human reproduction (Oxford, England)Nephropathic cystinosis presenting with uveitis: Report of a "Can't See, Can't Pee" situation.
Indian journal of pathology & microbiologyUnrecognized juvenile nephropathic cystinosis.
Kidney internationalLatest Clinical Approaches in the Ocular Management of Cystinosis: A Review of Current Practice and Opinion from the Ophthalmology Cystinosis Forum.
Ophthalmology and therapyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Cistinose nefropática juvenil.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Cistinose nefropática juvenil
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Multinucleated podocytes as a clue to diagnosis of juvenile nephropathic cystinosis.
- The Pitfall of White Blood Cell Cystine Measurement to Diagnose Juvenile Cystinosis.
- More than tubular dysfunction: cystinosis and kidney outcomes.
- Testicular function in males with infantile nephropathic cystinosis.
- Nephropathic cystinosis presenting with uveitis: Report of a "Can't See, Can't Pee" situation.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:411634(Orphanet)
- OMIM OMIM:219900(OMIM)
- MONDO:0009066(MONDO)
- GARD:17685(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55998574(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
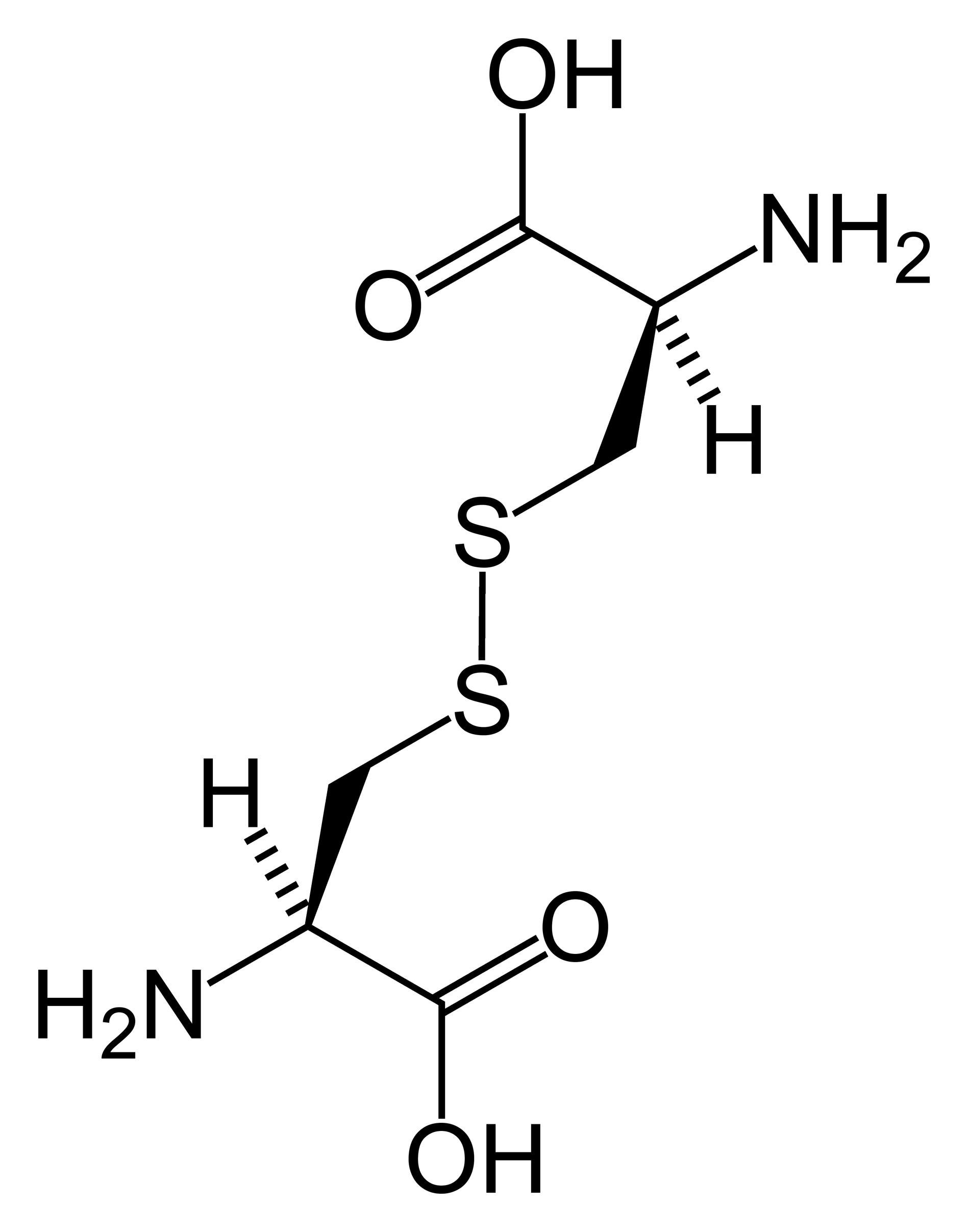
Cistinose nefropática juvenil
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub
- Ensaios clínicos
- fonte: ClinicalTrials.gov