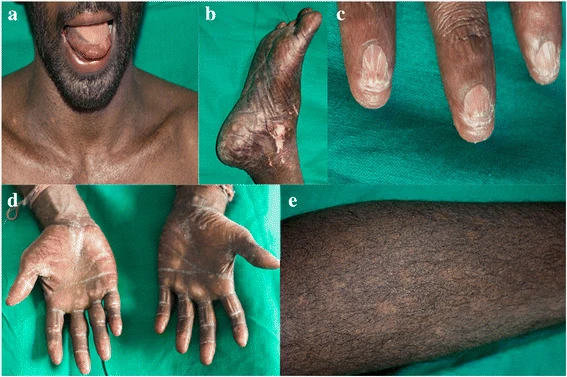
A disqueratose congênita (DC) é uma displasia ectodérmica rara que frequentemente se apresenta com a tríade clássica de displasia ungueal, alterações pigmentares da pele e leucoplasia oral associada a um alto risco de insuficiência da medula óssea (BMF) e câncer.
Introdução
O que você precisa saber de cara
A disqueratose congênita (DC) é uma displasia ectodérmica rara que frequentemente se apresenta com a tríade clássica de displasia ungueal, alterações pigmentares da pele e leucoplasia oral associada a um alto risco de insuficiência da medula óssea (BMF) e câncer.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 62 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 185 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
15 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, X-linked recessive.
Curadoria gene-doença
fontes oficiaisComponent of the shelterin complex (telosome) that is involved in the regulation of telomere length and protection. Shelterin associates with arrays of double-stranded TTAGGG repeats added by telomerase and protects chromosome ends; without its protective activity, telomeres are no longer hidden from the DNA damage surveillance and chromosome ends are inappropriately processed by DNA repair pathways. Plays a role in shelterin complex assembly. Isoform 1 may have additional role in tethering telo
NucleusChromosome, telomereNucleus matrix
Dyskeratosis congenita, autosomal dominant, 3
A rare multisystem disorder caused by defective telomere maintenance. It is characterized by progressive bone marrow failure, and the clinical triad of reticulated skin hyperpigmentation, nail dystrophy, and mucosal leukoplakia. Common but variable features include premature graying, aplastic anemia, low platelets, osteoporosis, pulmonary fibrosis, and liver fibrosis among others. Early mortality is often associated with bone marrow failure, infections, fatal pulmonary complications, or malignancy.
Component of the shelterin complex (telosome) that is involved in the regulation of telomere length and protection. Shelterin associates with arrays of double-stranded TTAGGG repeats added by telomerase and protects chromosome ends. Without its protective activity, telomeres are no longer hidden from the DNA damage surveillance and chromosome ends are inappropriately processed by DNA repair pathways. Promotes binding of POT1 to single-stranded telomeric DNA. Modulates the inhibitory effects of P
NucleusChromosome, telomere
Dyskeratosis congenita, autosomal dominant, 6
A form of dyskeratosis congenita, a rare multisystem disorder caused by defective telomere maintenance. It is characterized by progressive bone marrow failure, and the clinical triad of reticulated skin hyperpigmentation, nail dystrophy, and mucosal leukoplakia. Common but variable features include premature graying, aplastic anemia, low platelets, osteoporosis, pulmonary fibrosis, and liver fibrosis among others. Early mortality is often associated with bone marrow failure, infections, fatal pulmonary complications, or malignancy.
5'-3' exonuclease that plays a central role in telomere maintenance and protection during S-phase. Participates in the protection of telomeres against non-homologous end-joining (NHEJ)-mediated repair, thereby ensuring that telomeres do not fuse. Plays a key role in telomeric loop (T loop) formation by being recruited by TERF2 at the leading end telomeres and by processing leading-end telomeres immediately after their replication via its exonuclease activity: generates 3' single-stranded overhan
Chromosome, telomereNucleusCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Dyskeratosis congenita, autosomal recessive, 8
A form of dyskeratosis congenita, a rare multisystem disorder caused by defective telomere maintenance. It is characterized by progressive bone marrow failure, and the clinical triad of reticulated skin hyperpigmentation, nail dystrophy, and mucosal leukoplakia. Common but variable features include premature graying, aplastic anemia, low platelets, osteoporosis, pulmonary fibrosis, and liver fibrosis among others. Early mortality is often associated with bone marrow failure, infections, fatal pulmonary complications, or malignancy. Additional DKCB8 features include microcephaly, intrauterine growth retardation, and developmental anomalies in some patients. DKCB8 patients exhibit normal global telemore length, although there is evidence of telomere instability.
Required for ribosome biogenesis and telomere maintenance. Part of the H/ACA small nucleolar ribonucleoprotein (H/ACA snoRNP) complex, which catalyzes pseudouridylation of rRNA (PubMed:32554502). This involves the isomerization of uridine such that the ribose is subsequently attached to C5, instead of the normal N1. Each rRNA can contain up to 100 pseudouridine ('psi') residues, which may serve to stabilize the conformation of rRNAs. May also be required for correct processing or intranuclear tr
Nucleus, nucleolusNucleus, Cajal body
Dyskeratosis congenita, autosomal recessive, 1
A rare multisystem disorder caused by defective telomere maintenance. It is characterized by progressive bone marrow failure, and the clinical triad of reticulated skin hyperpigmentation, nail dystrophy, and mucosal leukoplakia. Common but variable features include premature graying, aplastic anemia, low platelets, osteoporosis, pulmonary fibrosis, and liver fibrosis among others. Early mortality is often associated with bone marrow failure, infections, fatal pulmonary complications, or malignancy.
Catalyzes the reductive methylation of 2'-deoxyuridine 5'-monophosphate (dUMP) to thymidine 5'-monophosphate (dTMP), using the cosubstrate, 5,10- methylenetetrahydrofolate (CH2H4folate) as a 1-carbon donor and reductant and contributes to the mitochondrial and nuclear de novo thymidylate biosynthesis pathway
NucleusCytoplasmMitochondrionMitochondrion matrixMitochondrion inner membrane
Dyskeratosis congenita, digenic
A form of dyskeratosis congenita, a rare multisystem disorder caused by defective telomere maintenance. It is characterized by progressive bone marrow failure, and the clinical triad of reticulated skin hyperpigmentation, nail dystrophy, and mucosal leukoplakia. Common but variable features include premature graying, aplastic anemia, low platelets, osteoporosis, pulmonary fibrosis, and liver fibrosis among others. Early mortality is often associated with bone marrow failure, infections, fatal pulmonary complications, or malignancy. DKCD transmission pattern is consistent with digenic inheritance.
Required for ribosome biogenesis and telomere maintenance. Part of the H/ACA small nucleolar ribonucleoprotein (H/ACA snoRNP) complex, which catalyzes pseudouridylation of rRNA. This involves the isomerization of uridine such that the ribose is subsequently attached to C5, instead of the normal N1. Each rRNA can contain up to 100 pseudouridine ('psi') residues, which may serve to stabilize the conformation of rRNAs. May also be required for correct processing or intranuclear trafficking of TERC,
Nucleus, nucleolusNucleus, Cajal body
Dyskeratosis congenita, autosomal recessive, 2
A rare multisystem disorder caused by defective telomere maintenance. It is characterized by progressive bone marrow failure, and the clinical triad of reticulated skin hyperpigmentation, nail dystrophy, and mucosal leukoplakia. Common but variable features include premature graying, aplastic anemia, low platelets, osteoporosis, pulmonary fibrosis, and liver fibrosis among others. Early mortality is often associated with bone marrow failure, infections, fatal pulmonary complications, or malignancy.
RNA chaperone that plays a key role in telomere maintenance and RNA localization to Cajal bodies (PubMed:29695869, PubMed:29804836). Specifically recognizes and binds the Cajal body box (CAB box) present in both small Cajal body RNAs (scaRNAs) and telomerase RNA template component (TERC) (PubMed:19285445, PubMed:20351177, PubMed:29695869, PubMed:29804836). Essential component of the telomerase holoenzyme complex, a ribonucleoprotein complex essential for the replication of chromosome termini tha
Nucleus, Cajal bodyChromosome, telomereChromosome
Dyskeratosis congenita, autosomal recessive, 3
A rare multisystem disorder caused by defective telomere maintenance. It is characterized by progressive bone marrow failure, and the clinical triad of reticulated skin hyperpigmentation, nail dystrophy, and mucosal leukoplakia. Common but variable features include premature graying, aplastic anemia, low platelets, osteoporosis, pulmonary fibrosis, and liver fibrosis among others. Early mortality is often associated with bone marrow failure, infections, fatal pulmonary complications, or malignancy.
3'-5' RNA exonuclease that trims the 3' end of oligo(U) and oligo(A) tracts of the pre-U6 small nuclear RNA (snRNA) molecule, leading to the formation of a mature U6 snRNA 3' end-terminated with a 2',3'-cyclic phosphate (PubMed:22899009, PubMed:23022480, PubMed:23190533, PubMed:26213367, PubMed:28887445, PubMed:30215753, PubMed:31832688). Participates in the U6 snRNA 3' end processing that prevents U6 snRNA degradation (PubMed:22899009, PubMed:23022480, PubMed:23190533, PubMed:26213367, PubMed:2
Nucleus
Poikiloderma with neutropenia
A genodermatosis characterized by poikiloderma, pachyonychia and chronic neutropenia. The disorder starts as a papular erythematous rash on the limbs during the first year of life. It gradually spreads centripetally and, as the papular rash resolves, hypo- and hyperpigmentation result, with development of telangiectasias. Another skin manifestation is pachyonychia, but alopecia and leukoplakia are distinctively absent. Patients have recurrent pneumonias that usually result in reactive airway disease and/or chronic cough. One of the most important extracutaneous symptoms is an increased susceptibility to infections, mainly affecting the respiratory system, primarily due to a chronic neutropenia and to neutrophil functional defects. Bone marrow abnormalities account for neutropenia and may evolve into myelodysplasia associated with the risk of leukemic transformation. Poikiloderma with neutropenia shows phenotypic overlap with Rothmund-Thomson syndrome.
Component of the CST complex proposed to act as a specialized replication factor promoting DNA replication under conditions of replication stress or natural replication barriers such as the telomere duplex. The CST complex binds single-stranded DNA with high affinity in a sequence-independent manner, while isolated subunits bind DNA with low affinity by themselves. Initially the CST complex has been proposed to protect telomeres from DNA degradation (PubMed:19854130). However, the CST complex ha
NucleusChromosome, telomere
Cerebroretinal microangiopathy with calcifications and cysts 1
An autosomal recessive pleiomorphic disorder characterized primarily by intracranial calcifications, leukodystrophy, and brain cysts, resulting in spasticity, ataxia, dystonia, seizures, and cognitive decline. Patients also have retinal telangiectasia and exudates (Coats disease) as well as extraneurologic manifestations, including osteopenia with poor bone healing and a high risk of gastrointestinal bleeding and portal hypertension caused by vasculature ectasias in the stomach, small intestine, and liver. Some individuals also have hair, skin, and nail changes, as well as anemia and thrombocytopenia.
Involved in diverse cellular processes such as ribosome biogenesis, centrosome duplication, protein chaperoning, histone assembly, cell proliferation, and regulation of tumor suppressors p53/TP53 and ARF. Binds ribosome presumably to drive ribosome nuclear export. Associated with nucleolar ribonucleoprotein structures and bind single-stranded nucleic acids. Acts as a chaperonin for the core histones H3, H2B and H4. Stimulates APEX1 endonuclease activity on apurinic/apyrimidinic (AP) double-stran
Nucleus, nucleolusNucleus, nucleoplasmCytoplasm, cytoskeleton, microtubule organizing center, centrosome
A probable ATP-dependent DNA helicase implicated in telomere-length regulation, DNA repair and the maintenance of genomic stability. Acts as an anti-recombinase to counteract toxic recombination and limit crossover during meiosis. Regulates meiotic recombination and crossover homeostasis by physically dissociating strand invasion events and thereby promotes noncrossover repair by meiotic synthesis dependent strand annealing (SDSA) as well as disassembly of D loop recombination intermediates. Als
Nucleus
Dyskeratosis congenita, autosomal recessive, 5
A form of dyskeratosis congenita, a rare multisystem disorder caused by defective telomere maintenance. It is characterized by progressive bone marrow failure, and the clinical triad of reticulated skin hyperpigmentation, nail dystrophy, and mucosal leukoplakia. Common but variable features include premature graying, aplastic anemia, low platelets, osteoporosis, pulmonary fibrosis, and liver fibrosis among others. Early mortality is often associated with bone marrow failure, infections, fatal pulmonary complications, or malignancy. DKCB5 is characterized by onset of bone marrow failure and immunodeficiency in early childhood. Most patients also have growth and developmental delay and cerebellar hypoplasia, consistent with a clinical diagnosis of Hoyeraal-Hreidarsson syndrome.
Catalytic subunit of H/ACA small nucleolar ribonucleoprotein (H/ACA snoRNP) complex, which catalyzes pseudouridylation of rRNA (PubMed:25219674, PubMed:32554502). This involves the isomerization of uridine such that the ribose is subsequently attached to C5, instead of the normal N1 (PubMed:25219674). Each rRNA can contain up to 100 pseudouridine ('psi') residues, which may serve to stabilize the conformation of rRNAs. Required for ribosome biogenesis and telomere maintenance (PubMed:19179534, P
Nucleus, nucleolusNucleus, Cajal bodyCytoplasm
Dyskeratosis congenita, X-linked
A rare, progressive bone marrow failure syndrome characterized by the triad of reticulated skin hyperpigmentation, nail dystrophy, and mucosal leukoplakia. Early mortality is often associated with bone marrow failure, infections, fatal pulmonary complications, or malignancy.
3'-exoribonuclease that has a preference for poly(A) tails of mRNAs, thereby efficiently degrading poly(A) tails. Exonucleolytic degradation of the poly(A) tail is often the first step in the decay of eukaryotic mRNAs and is also used to silence certain maternal mRNAs translationally during oocyte maturation and early embryonic development. Interacts with both the 3'-end poly(A) tail and the 5'-end cap structure during degradation, the interaction with the cap structure being required for an eff
NucleusCytoplasmNucleus, nucleolus
Dyskeratosis congenita, autosomal recessive, 6
A form of dyskeratosis congenita, a rare multisystem disorder caused by defective telomere maintenance. It is characterized by progressive bone marrow failure, and the clinical triad of reticulated skin hyperpigmentation, nail dystrophy, and mucosal leukoplakia. Common but variable features include premature graying, aplastic anemia, low platelets, osteoporosis, pulmonary fibrosis, and liver fibrosis among others. Early mortality is often associated with bone marrow failure, infections, fatal pulmonary complications, or malignancy.
Telomerase is a ribonucleoprotein enzyme essential for the replication of chromosome termini in most eukaryotes. Active in progenitor and cancer cells. Inactive, or very low activity, in normal somatic cells. Catalytic component of the teleromerase holoenzyme complex whose main activity is the elongation of telomeres by acting as a reverse transcriptase that adds simple sequence repeats to chromosome ends by copying a template sequence within the RNA component of the enzyme. Catalyzes the RNA-de
Nucleus, nucleolusNucleus, nucleoplasmNucleusChromosome, telomereCytoplasmNucleus, PML body
Variantes genéticas (ClinVar)
197 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 12.239 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
37 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Disqueratose congênita
Centros de Referência SUS
24 centros habilitados pelo SUS para Disqueratose congênita
Centros para Disqueratose congênita
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
6 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
20 ensaios clínicos encontrados, 7 ativos.
Publicações mais relevantes
Monoallelic PARN mutation presenting as pancytopenia, hepatic fibrosis and idiopathic pulmonary fibrosis.
A woman in her late 20s presented with a 5-year history of progressive fatigue and generalised weakness. Examination revealed signs of premature ageing, anaemia, neuropathy and hepatosplenomegaly.Investigations showed pancytopenia, dimorphic anaemia, severe vitamin B12 deficiency, hepatic fibrosis and pulmonary fibrosis. Despite correction of nutritional deficiencies, the constellation of cytopenia, premature greying, osteoporosis, hepatic and pulmonary fibrosis raised suspicion of a genetic disorder. Clinical exome sequencing identified a monoallelic PARN missense variant (c.613T>C; p.Cys205Arg), classified as a Variant of Uncertain Significance. Germline pathogenic variants in PARN have been associated with dyskeratosis congenita, autosomal recessive 6 (DKCB6) and with telomere-related pulmonary fibrosis and/or bone marrow failure 4. The clinical-genetic correlation in this case supported a diagnosis of a telomere biology disorder (TBD). This case highlights the importance of considering TBDs in young adults with unexplained multisystem disease, even when classical mucocutaneous features are absent. Early recognition is crucial for guiding genetic counselling, surveillance and consideration of bone marrow transplantation in progressive cases.
RNA Analysis Uncovers Pathogenic PARN Variant in Dyskeratosis Congenita.
Dyskeratosis congenita (DC) is a rare genetic disorder caused by impaired telomere maintenance, leading to diverse clinical manifestations, including bone marrow failure, mucocutaneous abnormalities, and multi-organ dysfunction. Here, we report a 14-year-old male patient presenting with microcephaly, developmental delay, synostoses, cerebellar hypoplasia with ataxia, and an immunodeficiency condition, but lacking classical DC features such as nail dystrophy and skin hyperpigmentation. WGS revealed two variants in the PARN gene: a known pathogenic missense variant (c.1045C > T, p.Arg349Trp) and a novel intronic variant (c.178-28T > C). Functional RNA analysis demonstrated that the intronic variant disrupts the branch point sequence, leading to exon 4 skipping and nonsense-mediated decay (NMD) of a significant proportion of transcripts. This study confirms the pathogenicity of the intronic variant and underscores the importance of functional validation in interpreting noncoding variants, particularly in genetically heterogeneous disorders like DC. Our findings expand the molecular and phenotypic spectrum of PARN-related DC and highlight the utility of WGS reanalysis and RNA studies in resolving diagnostically challenging cases.
Avascular Necrosis and Minimal Trauma Fractures in Telomere Biology Disorders.
Avascular necrosis (AVN) and minimal trauma fractures (MTF) cause significant morbidity in patients with telomere biology disorders (TBDs). TBDs are associated with very high risks of bone marrow failure, pulmonary fibrosis, cancer, and many other complications due to pathogenic germline variants in genes essential for telomere function and maintenance. To understand the extent to which AVN and MTF occur in TBDs and identify areas requiring more research in the role of telomeres in bone biology. We assessed the occurrence of AVN and MTF in 233 patients with TBDs. An age, gender, and gene-matched TBD patient control group was used to assess associations between AVN/MTF and clinical characteristics. Forty-two (18%) patients with TBD developed at least one AVN and/or MTF event with 19 patients experiencing their first event in childhood. AVN and MTF were most common in patients with autosomal or X-linked recessive, or heterozygous TINF2 disease (19/36 AVN and 17/19 MTF). Androgen and corticosteroid use were more common in patients with AVN compared with matched patient controls (41.2% vs. 16.3%, p < 0.05 and 41.2% vs. 14%, p < 0.01, respectively); however, 57.1% of patients experienced AVN and/or MTF events in the absence of androgen or corticosteroid use. Severe bone marrow failure and hematopoietic cell transplantation history were significantly more common in MTF patients than in controls (44.2% and 30.2% respectively, p < 0.05). There were no statistically significant associations between low bone mineral density or vitamin D deficiency and AVN or MTF. AVN and MTFs are common, debilitating complications in TBDs and frequently occur independently of androgen or corticosteroid use. Our results underscore the need for disease-specific translational studies as well as improved prevention and therapeutic options for patients with TBDs. Trial Registration: ClinicalTrials.gov identifier: NCT00027274.
Dyskerin dysfunction in cancer development: from telomere dysregulation to immune deficiency.
Dyskerin, encoded by the dyskerin pseudouridine synthase 1 (DKC1) gene, is a core component of the H/ACA ribonucleoprotein complex and plays essential roles in telomerase activity maintenance, rRNA pseudouridylation, and ribosome biogenesis. Loss of DKC1 function represents a major pathogenic basis of dyskeratosis congenita (DC) and is associated with a markedly increased risk of malignancy, particularly head and neck squamous cell carcinoma and oral squamous cell carcinoma. Traditionally, cancer susceptibility in DC has been largely attributed to telomere shortening and the resulting genomic instability; however, this explanation does not fully account for the heterogeneity observed across different genetic subtypes and clinical phenotypes. In this review, we systematically integrate three key mechanisms through which dyskerin dysfunction contributes to DC-associated carcinogenesis: disruption of telomere homeostasis, defects in selective translation regulation dependent on RNA pseudouridylation, and progressive impairment of T-cell-mediated immune surveillance. We highlight how DKC1 deficiency leads to insufficient rRNA pseudouridylation, selectively affecting the translation of internal ribosome entry site (IRES)-dependent transcripts, thereby attenuating the stress-induced expression of critical tumor suppressor proteins. In parallel, evidence from patient cohort studies is discussed to support a potentially dominant role of immunodeficiency in tumor development. Finally, we propose that future studies on DC and short telomere syndromes should emphasize genetic stratification and long-term clinical outcomes to refine cancer risk assessment and optimize preventive and therapeutic strategies.
Beyond iron deficiency: A comprehensive national survey of anaemia etiology in Sri Lankan young adults.
Previous studies in Sri Lanka have explored the prevalence and causes of anaemia, mainly emphasizing iron deficiency while overlooking other important factors such as enzymopathies, membranopathies, and haemoglobinopathies. Moreover, many studies reported unexplained cases of anaemia even after detailed evaluation. This study aimed to determine the prevalence and underlying causes of anaemia among community-based young adults in Sri Lanka. A descriptive cross-sectional study was conducted from January 2023 to December 2024 among young adults aged 18-30 years. Data and blood samples were collected using a pretested questionnaire. Anaemic individuals underwent serum iron studies, CRP, vitamin B12 and folate assays, and thalassaemia screening. Those with uncharacterized anaemia were further assessed using red cell enzyme assays, EMA dye binding assays, and whole-exome sequencing. The mean (± SD) age of the study participants was 23.90 ± 1.98 years. Anaemia prevalence was 15.0%. The main causes were iron deficiency (49.3%), folate deficiency (27.8%), vitamin B12 deficiency (14.4%), and haemoglobinopathy traits were identified in 25.6% of anaemic individuals, frequently in combination with nutritional deficiencies. A substantial proportion of anaemic individuals exhibited coexisting aetiologies rather than a single cause. Initially, 17.4% remained uncharacterized, but advanced testing identified variants suggestive of hereditary spherocytosis and congenital dyserythropoietic anaemia or dyskeratosis congenita. This first community-based Sri Lankan study integrating advanced diagnostics revealed that, although iron deficiency predominates, genetic and enzymatic disorders contribute notably, highlighting the need for broader diagnostic strategies in anaemia screening in the community.
Publicações recentes
Adriamycin, Vinblastine and Dacarbazine With Immunotherapy Achieves Complete Metabolic Response in a Patient With Classical Hodgkin Lymphoma and Dyskeratosis Congenita.
Late-onset dyskeratosis congenita due to a TERC (n.269G > C) variant-first reported case from Indonesia: a case report.
Rare somatic manifestations of telomere biology disorders.
Bone marrow failure in telomere biology disorders: Current understanding and the emerging landscape of non-transplant therapies.
Clonal hematopoiesis in telomere biology disorders.
📚 EuropePMC699 artigos no totalmostrando 195
Dyskerin dysfunction in cancer development: from telomere dysregulation to immune deficiency.
American journal of cancer researchBeyond iron deficiency: A comprehensive national survey of anaemia etiology in Sri Lankan young adults.
Scientific reportsDyskeratosis Congenita: Clinical Phenotype and Genetic Features in a Sibling Pair.
Clinical, cosmetic and investigational dermatologyMonoallelic PARN mutation presenting as pancytopenia, hepatic fibrosis and idiopathic pulmonary fibrosis.
BMJ case reportsDyskeratosis Congenita Due to a de novo TINF2 Mutation.
QJM : monthly journal of the Association of PhysiciansCirrhosis of Liver in Patients With Dyskeratosis Congenita: A Report of Two Cases.
Clinical case reportsExosome-mediated decay of unstable long extended precursors of human telomerase RNA is dependent on 5'-cap trimethylation.
Genes & developmentA rare case of dyskeratosis congenita with DKC1 mutation presenting initially as thrombocytopenia: Case report.
MedicineOral squamous cell carcinoma risk and magnitude of association in inherited cancer predisposition syndromes: evidence from a large real-world cohort.
Oral surgery, oral medicine, oral pathology and oral radiology[Clinical and genetic analysis of a child with X-linked Hoyeraal-Hreidarsson syndrome due to variant of DKC1 gene and a literature review].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsRecent progress in human telomerase structure and its therapeutic targeting.
Frontiers in molecular biosciencesInterprotomer communication and functional asymmetry in H/ACA snoRNPs.
Proceedings of the National Academy of Sciences of the United States of AmericaCase Report of Hair Abnormalities in Dyskeratosis Congenita: A Trichoscopic and Microscopic Analysis.
Skin appendage disordersFueling for the finish line: Control of human telomerase activity by nucleotide metabolism.
DNA repairCase Series: Clinical Significance of Heterozygous Pathogenic RTEL1 Variants Identified via Routine Clinical Genetic Diagnostics.
American journal of medical genetics. Part A[Clinical and genetic characteristics of 6 cases of congenital dyskeratosis in children].
Zhonghua er ke za zhi = Chinese journal of pediatricsSegmental dyskeratosis congenita - A diagnostic challenge.
Anais brasileiros de dermatologiaBeyond Hematologic Malignancies: Colorectal Cancer as a Solid Tumor Manifestation of Inherited Bone Marrow Failure Syndromes.
International journal of molecular sciencesEnding diagnostic odyssey by reanalysis of whole exome sequencing data: reclassification of suspected Fanconi anemia cases to dyskeratosis congenita and Diamond-Blackfan anemia.
Orphanet journal of rare diseasesInsights in bone marrow failure syndromes: take home messages from the 3rd ESH-EBMT-EHA-IPIG translational research conference.
Bone marrow transplantationOptical Genomic Mapping and Next-Generation Sequencing Identified Retrotransposon Insertion and Missense Variant Disrupting PARN Gene in Dyskeratosis Congenita.
Human mutationRNA Analysis Uncovers Pathogenic PARN Variant in Dyskeratosis Congenita.
Clinical geneticsTelomerase activity in T-cells as a functional test for pathogenicity assessment of novel genetic variants in telomere biology disorders.
Scientific reportsAvascular Necrosis and Minimal Trauma Fractures in Telomere Biology Disorders.
Clinical geneticsTargeting DKC1/NF-κB axis suppresses tumorigenesis and enhances 5-FU sensitivity in gastric cancer.
Cancer cell internationalSevere Treatment-Related Toxicity in Oral Cavity Squamous Cell Carcinoma with Dyskeratosis Congenita: A Case Report and Critical Review of Radiation-Induced Complications.
Advances in radiation oncologyThe role of telomeres in leukemic stem cells function.
Regenerative therapyIdentification and Computational Analysis of a Novel Pathogenic DKC1 Variant Underlying X-Linked Dyskeratosis Congenita.
Biochemical genetics[Clinical Analysis of Dyskeratosis Congenita in Children].
Zhongguo shi yan xue ye xue za zhiIntronic hexanucleotide repeat expansion in TYMS in monozygotic twins with congenital progressive universal melanosis.
Biomedical reportsMultidisciplinary Approach to Familial Pulmonary Fibrosis.
Radiographics : a review publication of the Radiological Society of North America, IncBilateral, recurrent Cytomegalovirus retinitis in Dyskeratosis congenita healing with extensive dystrophic calcification.
International ophthalmologySeparation of telomere protection from length regulation by two different point mutations at amino acid 492 of RTEL1.
Nucleic acids research[Dyskeratosis congenita combined with myeloproliferative disorder and trilineage cytopenia].
Zhonghua jie he he hu xi za zhi = Zhonghua jiehe he huxi zazhi = Chinese journal of tuberculosis and respiratory diseasesInvestigating telomere length in progeroid syndromes: implications for aging disorders.
AgingGermline PARN Variants in Telomere Biology Disorders and Challenges in Variant Curation.
Molecular genetics & genomic medicineAnalysis of Late Complications Associated With Hematopoietic Stem Cell Transplantation in Patients With Dyskeratosis Congenita.
Pediatric blood & cancerSiblings with a Homozygous Variant in the NHP2 Gene: A Case Report and Review of Literature.
Molecular syndromologyDe Novo Splice-Site Variant in DKC1 in a Female With Clinical Features of Hoyeraal-Hreidarsson Syndrome.
American journal of medical genetics. Part AExtensive and persistent tongue ulceration is an early character of dyskeratosis congenita.
Orphanet journal of rare diseasesStructural Biology of Telomerase and Associated Factors.
Cold Spring Harbor perspectives in biologyLoss of Ten1 in mice induces telomere shortening and models human dyskeratosis congenita.
Science advancesTYMS-ENOSF1 Dyskeratosis Congenita in a Patient With Ring Chromosome 18: A Case Report.
American journal of medical genetics. Part AFamilial pulmonary fibrosis with dyskeratosis congenita associated with a rare RTEL1 gene mutation.
BMJ case reportsDyskeratosis Congenita Complicated by Pulmonary Fibrosis and Myelodysplastic Syndrome with a Germline Mutation of the DKC1 Gene and a Somatic Mutation of the U2AF1 Gene in Leukocytes.
Internal medicine (Tokyo, Japan)Expanding the clinical spectrum of NHP2 -related dyskeratosis congenita: a case with novel phenotypic features.
Clinical dysmorphologyLinear hypermelanosis in narrow bands: a mosaic pattern manifestation of dyskeratosis congenita?
The British journal of dermatologyHaploidentical Hematopoietic Cell Transplantation in Dyskeratosis Congenita with Myelodysplastic Syndrome/Acute Myeloid Leukemia.
Blood cell therapyGenetic variants in NHEJ1 and related DNA repair disorders: insights into phenotypic heterogeneity and links to hypoplastic myelodysplastic syndromes and familial hematological malignancies susceptibility.
Annals of hematologyClinical Use of ZSCAN4 for Telomere Elongation in Hematopoietic Stem Cells.
NEJM evidencePremature ageing of lung alveoli and bone marrow cells from Terc deficient mice with different telomere lengths.
Scientific reportsPhenotype puzzle: the role of novel LMBRD1 gene variant in Cbl deficiency causing Dyskeratosis Congenita-like clinical manifestations.
Journal of human geneticsLate-onset telomere biology disorders in adults: clinical insights and treatment outcomes from a retrospective registry cohort.
Blood advancesDyskeratosis congenita mistaken for lichen planus.
Archives of disease in childhoodTERT de novo mutation-associated dyskeratosis congenita and porto-sinusoidal vascular disease: a case report.
Journal of medical case reports[Next generation sequencing in pediatric bone marrow failure: a valuable tool for accurate diagnosis].
Andes pediatrica : revista Chilena de pediatriaBilateral cytomegalovirus retinitis in a patient with dyskeratosis congenita.
International journal of ophthalmology"Crying in the Wilderness"-The Use of Web-Based Support in Telomere Biology Disorders: Thematic Analysis.
JMIR formative researchHoyeraal-Hreidarsson syndrome: a case report of dyskeratosis congenita with a novel PARN gene mutation.
Annals of medicine and surgery (2012)Telomere function and regulation from mouse models to human ageing and disease.
Nature reviews. Molecular cell biologyA "rotating menu" of medical uncertainty for families affected by telomere biology disorders: A qualitative interview study.
SSM. Qualitative research in healthInducible pluripotent stem cell models to study bone marrow failure and MDS predisposition syndromes.
Experimental hematologyInherited Telomere Biology Disorders: Pathophysiology, Clinical Presentation, Diagnostics, and Treatment.
Transfusion medicine and hemotherapy : offizielles Organ der Deutschen Gesellschaft fur Transfusionsmedizin und ImmunhamatologieTelomeres and immunodeficiencies.
Human immunologyAn Atypical Presentation of Dyskeratosis Congenita in a Child With a Familial RTEL1 Mutation.
Pediatric dermatologyGermline RTEL1 Variants in Telomere Biology Disorders.
American journal of medical genetics. Part AA comprehensive in silico investigation into the pathogenic SNPs in the RTEL1 gene and their biological consequences.
PloS oneLocally advanced rectal cancer in a young adult affected with dyskeratosis congenita (Zinsser-Cole-Engman syndrome): a case report.
Surgical case reportsReticulated pigmentary changes and Terry's nails in a patient with a TERT variant-associated telomere biology disorder.
Pediatric dermatologyThe gray boundaries of aberrant shortening of the cellular timekeepers' edges.
EMBO molecular medicineThe evolving genetic landscape of telomere biology disorder dyskeratosis congenita.
EMBO molecular medicineTelomere biology disorders: from dyskeratosis congenita and beyond.
Postgraduate medical journalPresumptive Cytomegalovirus Retinitis as a Complication of Dyskeratosis Congenita: A Case Report.
Case reports in ophthalmologyAn algorithmic approach towards diagnosis of patients with hereditary reticulate pigmentary disorders: a narrative review.
Clinical and experimental dermatologyMucosal Cancers Arising in Potentially Malignant Lesions of the Oral Mucosa Are Marjolin Ulcers: New Insights Into Old Concepts.
Dermatology practical & conceptualNucleotide sugars correlate with leukocyte telomere length as part of a dyskeratosis congenita metabolomic plasma signature.
HaematologicaA case of dermatopathia pigmentosa reticularis masquerading as dyskeratosis congenita: the importance of nailing the correct diagnosis.
Clinical and experimental dermatologyDiseases with oral malignant potential: Need for change to inform research, policy, and practice.
Journal of oral pathology & medicine : official publication of the International Association of Oral Pathologists and the American Academy of Oral PathologyA Novel Autosomal Recessive Candidate Gene Responsible for RASopathy-Like Phenotype and Bone Marrow Failure: RASA3.
Journal of pediatric geneticsBilateral retinal vasculopathy in a patient with aplastic anemia due to dyskeratosis congenita.
Journal francais d'ophtalmologieMASCC/ISOO Clinical Practice Statement: The risk of secondary oral cancer following hematopoietic cell transplantation.
Supportive care in cancer : official journal of the Multinational Association of Supportive Care in CancerReduced anti-Müllerian hormone levels in males with inherited bone marrow failure syndromes.
Endocrine connectionsDyskeratosis congenita with squamous cell carcinoma of the tongue: A rare case report.
Indian journal of pathology & microbiologyAllogeneic Hematopoietic Cell Transplant For Bone Marrow Failure or Myelodysplastic Syndrome in Dyskeratosis Congenita/Telomere Biology Disorders: Single-Center, Single-Arm, Open-Label Trial of Reduced-Intensity Conditioning Without Radiation.
Transplantation and cellular therapyDyskeratosis congenita associated with a novel missense variant in TERT: Approach for the dermatologists.
Archives of dermatological researchCongenital nail abnormalities.
Hand surgery & rehabilitationGermline thymidylate synthase deficiency impacts nucleotide metabolism and causes dyskeratosis congenita.
American journal of human geneticsOverexpression of human SAMD9 inhibits protein translation and alters MYC signaling resulting in cell cycle arrest.
Experimental hematologySkin cancer-associated genodermatoses in skin of color patients: a review.
Archives of dermatological researchDyskeratosis congenita: a rare case report.
Oxford medical case reportsIntegrated proteogenomic analysis for inherited bone marrow failure syndrome.
LeukemiaCancer Precursor Syndromes and Their Detection in the Head and Neck.
Hematology/oncology clinics of North AmericaA naturally occurring canine model of syndromic congenital microphthalmia.
G3 (Bethesda, Md.)Clinical mutations in the TERT and TERC genes coding for telomerase components induced oxidative stress, DNA damage at telomeres and cell apoptosis besides decreased telomerase activity.
Human molecular geneticsA Review of the Repair of DNA Double Strand Breaks in the Development of Oral Cancer.
International journal of molecular sciencesMultiple-Digit Pigmented Bowen's Disease Induced by Human Papillomavirus in an Immunocompetent Child.
Skin appendage disordersX-linked Dyskeratosis Congenita Case with Mutation 1058C>T(p.Ala353Val) in Dyskerine Gene.
Indian journal of dermatologyLiver involvement in dyskeratosis congenita-Case series and concise literature review.
Indian journal of gastroenterology : official journal of the Indian Society of GastroenterologyCan telomeric changes orchestrate the development of autoinflammatory skin diseases?
Italian journal of dermatology and venereologyA new variant in the ZCCHC8 gene: diverse clinical phenotypes and expression in the lung.
ERJ open researchLinking Gene Fusions to Bone Marrow Failure and Malignant Transformation in Dyskeratosis Congenita.
International journal of molecular sciencesInherited bone marrow failure syndromes: phenotype as a tool for early diagnostic suspicion at a major reference center in Mexico.
Frontiers in geneticsThe Use of Social Media to Express and Manage Medical Uncertainty in Dyskeratosis Congenita: Content Analysis.
JMIR infodemiologyCharacterization of novel mutations in the TEL-patch domain of the telomeric factor TPP1 associated with telomere biology disorders.
Human molecular geneticsPrevalence of Oral Submucous Fibrosis With Other Oral Potentially Malignant Disorders: A Clinical Retrospective Study.
CureusTelomere length and cancer risk: finding Goldilocks.
BiogerontologyA pan-cancer analysis of Dyskeratosis congenita 1 (DKC1) as a prognostic biomarker.
HereditasDigital telomere measurement by long-read sequencing distinguishes healthy aging from disease.
bioRxiv : the preprint server for biologyMinimal intensity conditioning strategies for bone marrow failure: is it time for "preventative" transplants?
Hematology. American Society of Hematology. Education ProgramPosttransplant complications in patients with marrow failure syndromes: are we improving long-term outcomes?
Hematology. American Society of Hematology. Education ProgramThe metabolic basis of inherited neutropenias.
British journal of haematologyCytogenetics in the management of bone marrow failure syndromes: Guidelines from the Groupe Francophone de Cytogénétique Hématologique (GFCH).
Current research in translational medicineX-linked genodermatoses from diagnosis to tailored therapy.
La Clinica terapeuticaTelomere biology disorders may manifest as common variable immunodeficiency (CVID).
Clinical immunology (Orlando, Fla.)Conformational plasticity and allosteric communication networks explain Shelterin protein TPP1 binding to human telomerase.
Communications chemistryA rare homozygous variant in TERT gene causing variable bone marrow failure, fragility fractures, rib anomalies and extremely short telomere lengths with high serum IgE.
British journal of haematologySevere Thrombocytopenia Due to Bone Marrow Failure in Children With Dyskeratosis Congenita Does Not Respond to Eltrombopag Treatment: Case Series.
Journal of pediatric hematology/oncologyThe C-terminal extension of dyskerin is a dyskeratosis congenita mutational hotspot that modulates interaction with telomerase RNA and subcellular localization.
Human molecular geneticsFyn-mediated phosphorylation of Menin disrupts telomere maintenance in stem cells.
bioRxiv : the preprint server for biologyGermline landscape of RPA1, RPA2 and RPA3 variants in pediatric malignancies: identification of RPA1 as a novel cancer predisposition candidate gene.
Frontiers in oncologyCompound heterozygous mutations in the helicase RTEL1 causing Hoyeraal-Hreidarsson syndrome with Blake`s pouch cyst: a case report.
The Turkish journal of pediatricsp53 in the Molecular Circuitry of Bone Marrow Failure Syndromes.
International journal of molecular sciencesGenetic Counseling and Family Screening Recommendations in Patients with Telomere Biology Disorders.
Current hematologic malignancy reportsNew Insights into Dyskerin-CypA Interaction: Implications for X-Linked Dyskeratosis Congenita and Beyond.
GenesTelomerase RNA-based aptamers restore defective myelopoiesis in congenital neutropenic syndromes.
Nature communicationsCRISPR screen identifies CEBPB as contributor to dyskeratosis congenita fibroblast senescence via augmented inflammatory gene response.
G3 (Bethesda, Md.)A case report of dyskeratosis congenita caused by a novel TERC mutation.
Annals of hematologyAC125611.3 promotes the progression of colon cancer by recruiting DKC1 to stabilize CTNNB1.
Arab journal of gastroenterology : the official publication of the Pan-Arab Association of GastroenterologyA systematic approach identifies p53-DREAM pathway target genes associated with blood or brain abnormalities.
Disease models & mechanismsThe Molecular and Genetic Mechanisms of Inherited Bone Marrow Failure Syndromes: The Role of Inflammatory Cytokines in Their Pathogenesis.
BiomoleculesDyskeratosis congenita: natural history of the disease through the study of a cohort of patients diagnosed in childhood.
Frontiers in pediatrics[Dyskeratosis congenita und Ösophagusstriktur bei einem Kleinkind].
Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDGThe PARN, TOE1, and USB1 RNA deadenylases and their roles in non-coding RNA regulation.
The Journal of biological chemistryInherited bone marrow failure syndromes and germline predisposition to myeloid neoplasia: A practical approach for the pathologist.
Seminars in diagnostic pathologyControl of protein synthesis through mRNA pseudouridylation by dyskerin.
Science advancesDyskeratosis Congenita: A Case Report of a Patient With Coronary Artery Disease.
CureusInherited Reticulate Pigmentary Disorders.
GenesThe distribution and accumulation of the shortest telomeres in telomere biology disorders.
British journal of haematologyPatient-Induced Pluripotent Stem Cell-Derived Hepatostellate Organoids Establish a Basis for Liver Pathologies in Telomeropathies.
Cellular and molecular gastroenterology and hepatologyHematopoietic cell transplantation for telomere biology diseases: A retrospective single-center cohort study.
European journal of haematologySpectrum of Liver Pathology in Dyskeratosis Congenita.
The American journal of surgical pathologyEndoscopic Assessment and Serial Balloon Dilatation in a Toddler With Dyskeratosis Congenita-Hoyeraal-Hreidarsson Syndrome Following Bone Marrow Transplant: A Case Report.
JPGN reportsWhat is the future of telomere length testing in telomere biology disorders?
Expert review of hematologySize matters in telomere biology disorders ‒ expanding phenotypic spectrum in patients with long or short telomeres.
Hereditary cancer in clinical practiceProgression of liver disease and portal hypertension in dyskeratosis congenita and related telomere biology disorders.
Hepatology (Baltimore, Md.)A Primary Gastrointestinal Presentation and Novel Genetic Variant of Dyskeratosis Congenita in a Pediatric Patient.
JPGN reportsBuildup from birth onward of short telomeres in human hematopoietic cells.
Aging cellIdentification of Adult Patients With Classical Dyskeratosis Congenita or Cryptic Telomere Biology Disorder by Telomere Length Screening Using Age-modified Criteria.
HemaSphereA de novo TINF2, R282C Mutation in a Case of Dyskeratosis Congenital Founded by Next-Generation Sequencing.
Iranian biomedical journalIdiopathic non-cirrhotic portal hypertension in dyskeratosis congenita with rare variant of NHP2.
QJM : monthly journal of the Association of PhysiciansCase report: A novel mutation in RTEL1 gene in dyskeratosis congenita.
Frontiers in oncologyBoosting NAD ameliorates hematopoietic impairment linked to short telomeres in vivo.
GeroScienceNeonatal intestinal obstruction in Hoyeraal-Hreidarsson syndrome with novel RTEL1 variants.
Pediatric blood & cancerDyskeratosis congenita with heterozygous RTEL1 mutations presenting with fibrotic hypersensitivity pneumonitis.
Respiratory medicine case reportsDyskeratosis Congenita Links Telomere Attrition to Age-Related Systemic Energetics.
The journals of gerontology. Series A, Biological sciences and medical sciencesGermline Pathogenic Variants in Squamous Cell Carcinoma of the Head and Neck.
Folia biologicaSubretinal drusenoid deposits and bilateral capillary peripheral occlusion in a patient with dyskeratosis congenita.
Canadian journal of ophthalmology. Journal canadien d'ophtalmologieAutoimmune Neutropenia and Immune-Dysregulation in a Patient Carrying a TINF2 Variant.
International journal of molecular sciencesDyskeratosis congenita and telomere biology disorders.
Hematology. American Society of Hematology. Education ProgramNovel TINF2 gene mutation in dyskeratosis congenita with extremely short telomeres: A case report.
World journal of clinical casesDermoscopic and Onychoscopic Features of Dyskeratosis Congenita.
Indian dermatology online journalRecent advances in understanding telomere diseases.
Faculty reviewsA missense variant in the nuclear localization signal of DKC1 causes Hoyeraal-Hreidarsson syndrome.
NPJ genomic medicineLate Presentation of Dyskeratosis Congenita: Germline Predisposition to Adult-Onset Secondary Acute Myeloid Leukemia.
Hematology reportsPainful thickened skin on the soles of the feet.
JAAD case reportsRegulator of telomere elongation helicase 1 gene and its association with malignancy.
Cancer reports (Hoboken, N.J.)de Novo TINF2 C.845G>A: Pathogenic Variant in Patient with Dyskeratosis Congenita.
Balkan journal of medical genetics : BJMGPreretinal Microvascular Tufts Associated with Dyskeratosis Congenita.
Ophthalmology[Bone marrow failure and TP53 activating mutations].
[Rinsho ketsueki] The Japanese journal of clinical hematologyCoats plus syndrome with new observation of drusenoid retinal pigment epithelial detachments in a teenager.
American journal of ophthalmology case reportsNext-generation sequencing errors due to genetic variation in WRAP53 encoding TCAB1 on chromosome 17.
Human mutationA Novel Variant and a Missense Variant Identified in the DKC1 Gene in Three Chinese Familieswith Dyskeratosis Congenita.
Clinical, cosmetic and investigational dermatologyFanconi anemia and dyskeratosis congenita/telomere biology disorders: Two inherited bone marrow failure syndromes with genomic instability.
Frontiers in oncologySevere immunochemotherapy-induced toxicities in a patient with dyskeratosis congenita and literature review.
Hematology (Amsterdam, Netherlands)Telomere biology disorders: time for moving towards the clinic?
Trends in molecular medicineDomain specific mutations in dyskerin disrupt 3' end processing of scaRNA13.
Nucleic acids researchThe biology and management of dyskeratosis congenita and related disorders of telomeres.
Expert review of hematologyChallenges in the interpretation of a germline TERT variant in a patient with juvenile myelomonocytic leukemia.
Pediatric blood & cancerPediatric cystectomy for refractory cystitis post-bone marrow transplant in dyskeratosis congenita: A case report.
Urology case reportsNAD-Linked Metabolism and Intervention in Short Telomere Syndromes and Murine Models of Telomere Dysfunction.
Frontiers in agingEffects of Recloning on the Telomere Lengths of Mouse Terc+/- Nuclear Transfer-Derived Embryonic Stem Cells.
Stem cells and developmentChemical Modifications of Ribosomal RNA.
Methods in molecular biology (Clifton, N.J.)Dysplastic megakaryocytes in dyskeratosis congenita with variant in PARN.
BloodSilencing circular RNA-friend leukemia virus integration 1 restrained malignancy of CC cells and oxaliplatin resistance by disturbing dyskeratosis congenita 1.
Open life sciencesRecent advances in hematopoietic cell transplantation for inherited bone marrow failure syndromes.
International journal of hematologyDyskerin Downregulation Can Induce ER Stress and Promote Autophagy via AKT-mTOR Signaling Deregulation.
BiomedicinesInherited bone marrow failure in the pediatric patient.
BloodTruncating TINF2 p.Tyr312Ter variant and inherited breast cancer susceptibility.
Familial cancerGSK3 inhibition rescues growth and telomere dysfunction in dyskeratosis congenita iPSC-derived type II alveolar epithelial cells.
eLifeGenetics and genomics of bone marrow failure syndrome.
Blood researchAre Dyskeratosis Congenita patients at higher risk of symptomatic COVID-19?
Medical hypothesesCase Report: Novel Biallelic Variants in DNAJC21 Causing an Inherited Bone Marrow Failure Spectrum Phenotype: An Odyssey to Diagnosis.
Frontiers in geneticsCase Report: A Missense Mutation in Dyskeratosis Congenita 1 Leads to a Benign Form of Dyskeratosis Congenita Syndrome With the Mucocutaneous Triad.
Frontiers in pediatricsSex differences in telomere length, lifespan, and embryonic dyskerin levels.
Aging cellEditing TINF2 as a potential therapeutic approach to restore telomere length in dyskeratosis congenita.
BloodAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Disqueratose congênita.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Disqueratose congênita
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Monoallelic PARN mutation presenting as pancytopenia, hepatic fibrosis and idiopathic pulmonary fibrosis.
- RNA Analysis Uncovers Pathogenic PARN Variant in Dyskeratosis Congenita.
- Avascular Necrosis and Minimal Trauma Fractures in Telomere Biology Disorders.
- Dyskerin dysfunction in cancer development: from telomere dysregulation to immune deficiency.
- Beyond iron deficiency: A comprehensive national survey of anaemia etiology in Sri Lankan young adults.
- Adriamycin, Vinblastine and Dacarbazine With Immunotherapy Achieves Complete Metabolic Response in a Patient With Classical Hodgkin Lymphoma and Dyskeratosis Congenita.
- Late-onset dyskeratosis congenita due to a TERC (n.269G > C) variant-first reported case from Indonesia: a case report.
- Rare somatic manifestations of telomere biology disorders.
- Bone marrow failure in telomere biology disorders: Current understanding and the emerging landscape of non-transplant therapies.
- Clonal hematopoiesis in telomere biology disorders.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1775(Orphanet)
- MONDO:0015780(MONDO)
- GARD:10905(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3709312(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Disqueratose congênita
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub
- Ensaios clínicos
- fonte: ClinicalTrials.gov