A Distrofia Muscular Congênita tipo 1B é uma doença neuromuscular genética e rara, caracterizada por fraqueza muscular que afeta ambos os lados do corpo de forma simétrica, especialmente nos músculos mais próximos ao tronco, como os do pescoço, rosto e diafragma (o principal músculo da respiração). Outras características incluem rigidez na coluna, contraturas nas articulações (dificuldade de movimento em áreas como o tendão de Aquiles, cotovelos e mãos), aumento generalizado dos músculos e insuficiência respiratória precoce (dificuldade grave para respirar), que geralmente se manifesta antes dos 10 anos de idade. Os pacientes costumam apresentar atraso nos marcos do desenvolvimento motor (como sentar, engatinhar e andar) e níveis muito elevados de creatina quinase (CK) no sangue, uma enzima que indica lesão muscular. Com o avanço da doença, podem surgir dificuldade para expelir o ar com força dos pulmões (usando os músculos da barriga) e respiração insuficiente durante o sono (hipoventilação noturna).
Introdução
O que você precisa saber de cara
A Distrofia Muscular Congênita tipo 1B é uma doença neuromuscular genética e rara, caracterizada por fraqueza muscular que afeta ambos os lados do corpo de forma simétrica, especialmente nos músculos mais próximos ao tronco, como os do pescoço, rosto e diafragma (o principal músculo da respiração). Outras características incluem rigidez na coluna, contraturas nas articulações (dificuldade de movimento em áreas como o tendão de Aquiles, cotovelos e mãos), aumento generalizado dos músculos e insuficiência respiratória precoce (dificuldade grave para respirar), que geralmente se manifesta antes dos 10 anos de idade. Os pacientes costumam apresentar atraso nos marcos do desenvolvimento motor (como sentar, engatinhar e andar) e níveis muito elevados de creatina quinase (CK) no sangue, uma enzima que indica lesão muscular. Com o avanço da doença, podem surgir dificuldade para expelir o ar com força dos pulmões (usando os músculos da barriga) e respiração insuficiente durante o sono (hipoventilação noturna).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 7 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 15 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Distrofia muscular congênita, tipo 1b
Centros de Referência SUS
24 centros habilitados pelo SUS para Distrofia muscular congênita, tipo 1b
Centros para Distrofia muscular congênita, tipo 1b
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
3 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Mostrando amostra de 6 publicações de um total de 636
Characterization of cardiac involvement in children with LMNA-related muscular dystrophy.
Introduction: LMNA-related muscular dystrophy is a rare entity that produce "laminopathies" such as Emery-Dreifuss muscular dystrophy (EDMD), limb-girdle muscular dystrophy type 1B (LGMD1B), and LMNA-related congenital muscular dystrophy (L-CMD). Heart failure, malignant arrhythmias, and sudden death may occur. No consensus exists on cardiovascular management in pediatric laminopathies. The aim was to perform an exhaustive cardiologic follow-up in pediatric patients diagnosed with LMNA-related muscular dystrophy. Methods: Baseline cardiac work-up consisted of clinical assessment, transthoracic Doppler echocardiography, 12-lead electrocardiogram, electrophysiological study, and implantation of a long-term implantable cardiac loop recorder (ILR). Results: We enrolled twenty-eight pediatric patients diagnosed with EDMD (13 patients), L-CMD (11 patients), LGMD1B (2 patients), and LMNA-related mild weakness (2 patients). Follow-up showed dilated cardiomyopathy (DCM) in six patients and malignant arrhythmias in five (four concomitant with DCM) detected by the ILR that required implantable cardioverter defibrillator (ICD) implantation. Malignant arrhythmias were detected in 20% of our cohort and early-onset EDMD showed worse cardiac prognosis. Discussion: Patients diagnosed with early-onset EDMD are at higher risk of DCM, while potentially life-threatening arrhythmias without DCM appear earlier in L-CMD patients. Early onset neurologic symptoms could be related with worse cardiac prognosis. Specific clinical guidelines for children are needed to prevent sudden death.
Clinical spectrum and genetic variations of LMNA-related muscular dystrophies in a large cohort of Chinese patients.
LMNA-related muscular dystrophy is caused by mutations in LMNA gene. We aimed to identify genetic variations and clinical features in a large cohort of Chinese patients with LMNA mutations in an attempt to establish genotype-phenotype correlation. The clinical presentations of patients with LMNA-related muscular dystrophy were recorded using retrospective and prospective cohort study. LMNA mutation analysis was performed by Sanger sequencing or next-generation sequencing. Mosaicism was detected by personal genome machine amplicon deep sequencing for mosaicism. Eighty-four patients were identified to harbour LMNA mutations. Forty-one of those were diagnosed with LMNA-related congenital muscular dystrophy (L-CMD), 32 with Emery-Dreifuss muscular dystrophy (EDMD) and 11 with limb-girdle muscular dystrophy type 1B (LGMD1B). We identified 21 novel and 29 known LMNA mutations. Two frequent mutations were identified: c.745C>T and c.1357C>T. A correlation between the location of mutation and the clinical phenotype was observed: mutations affecting the head and coil 2A domains mainly occurred in L-CMD, while the coil 2B and Ig-like domains mainly related to EDMD and LGMD1B. We found somatic mosaicism in one parent of four probands. Muscle biopsies revealed 11 of 20 biopsied L-CMD exhibited inflammatory changes, and muscle cell ultrastructure showed abnormal nuclear morphology. Our detailed clinical and genetic analysis of 84 patients with LMNA-related muscular dystrophy expands clinical spectrum and broadens genetic variations caused by LMNA mutations. We identified 21 novel and 29 known LMNA mutations and found two frequent mutations. A correlation between the location of mutation and the clinical severity was observed. Preliminary data suggested that low-dose corticosteroid treatment may be effective.
Lamin-Related Congenital Muscular Dystrophy Alters Mechanical Signaling and Skeletal Muscle Growth.
Laminopathies are a clinically heterogeneous group of disorders caused by mutations in the LMNA gene, which encodes the nuclear envelope proteins lamins A and C. The most frequent diseases associated with LMNA mutations are characterized by skeletal and cardiac involvement, and include autosomal dominant Emery-Dreifuss muscular dystrophy (EDMD), limb-girdle muscular dystrophy type 1B, and LMNA-related congenital muscular dystrophy (LMNA-CMD). Although the exact pathophysiological mechanisms responsible for LMNA-CMD are not yet understood, severe contracture and muscle atrophy suggest that mutations may impair skeletal muscle growth. Using human muscle stem cells (MuSCs) carrying LMNA-CMD mutations, we observe impaired myogenic fusion with disorganized cadherin/β catenin adhesion complexes. We show that skeletal muscle from Lmna-CMD mice is unable to hypertrophy in response to functional overload, due to defective fusion of activated MuSCs, defective protein synthesis and defective remodeling of the neuromuscular junction. Moreover, stretched myotubes and overloaded muscle fibers with LMNA-CMD mutations display aberrant mechanical regulation of the yes-associated protein (YAP). We also observe defects in MuSC activation and YAP signaling in muscle biopsies from LMNA-CMD patients. These phenotypes are not recapitulated in closely related but less severe EDMD models. In conclusion, combining studies in vitro, in vivo, and patient samples, we find that LMNA-CMD mutations interfere with mechanosignaling pathways in skeletal muscle, implicating A-type lamins in the regulation of skeletal muscle growth.
Cardiac diseases as a predictor warning of hereditary muscle diseases. The case of laminopathies.
Mutations in the LMNA gene are associated with a wide spectrum of disease phenotypes, ranging from neuromuscular, cardiac and metabolic disorders to premature aging syndromes. Skeletal muscle involvement may present with different phenotypes: limb-girdle muscular dystrophy type 1B or LMNA-related dystrophy; autosomal dominant Emery-Dreifuss muscular dystrophy; and a congenital form of muscular dystrophy, frequently associated with early onset of arrhythmias. Heart involvement may occur as part of the muscle involvement or independently, regardless of the presence of the myopathy. Notably conduction defects and dilated cardiomyopathy may exist without a muscle disease. This paper will focus on cardiac diseases presenting as the first manifestation of skeletal muscle hereditary disorders such as laminopathies, inspired by two large families with cardiovascular problems long followed by conventional cardiologists who did not suspect a genetic muscle disorder underlying these events. Furthermore it underlines the need for a multidisciplinary approach in these disorders and how the figure of the cardio-myo-geneticist may play a key role in facilitating the diagnostic process, and addressing the adoption of appropriate prevention measures.
Up-regulation of Toll-like receptors 7 and 9 and its potential implications in the pathogenic mechanisms of LMNA-related myopathies.
Laminopathies are a heterogeneous group of diseases, caused by mutations in lamin A/C proteins. The most common laminopathy (LMNA-related myopathies, LMNA-RM) affects skeletal and cardiac muscles; muscle histopathology is variable, ranging from mild unspecific changes to dystrophic features, sometimes with inflammatory evidence. Whether the genetic defect might activate innate immune components, leading to chronic inflammation, myofiber necrosis and fibrosis, is still unknown. By qPCR, a significant up-regulation of Toll-like receptor (TLR) 7 and 9 transcripts was found in LMNA-RM compared to other myopathic and non-myopathic muscles. A marked TLR7/9 staining was observed on LMNA-RM blood vessels and muscle fibers and, when present, on infiltrating cells, mainly macrophages, scattered in the tissue or localized close to degenerated muscle fibers and connective tissue. Our results recognize innate immunity as a player in LMNA-RM pathogenesis. Modulation of TLR7/9 signaling pathways and decrease of macrophage-mediated inflammation might be potential therapeutic strategies in LMNA-RM management. DMD, Duchenne muscular dystrophy; EDMD2, Emery-Dreifuss muscular dystrophy type 2; FSHD, facio-scapulo-humeral muscular dystrophy; LGMD1B, limb-girdle muscular dystrophy type 1B; LMNA-CMD, LMNA-related congenital muscular dystrophy; LMNA-RM, LMNA-related myopathies; sIBM, sporadic inclusion body myositis; TLR, Toll-like receptor.
Publicações recentes
Biosynthesis and Regulation of Laminin-Binding O-Mannosyl Glycans Related to Muscular Dystrophy.
Delayed presentation of muscular torticollis- an observational study.
Laminin-α2 is required for the maintenance of the myotendinous junction in vivo.
Clinico-genetic heterogeneity in Pakistani families affected with muscular dystrophies.
Pearls & Oy-sters: Use of Short-Acting B-Agonist in DOK7-Related Congenital Myasthenic Syndrome Treatment.
📚 EuropePMC943 artigos no totalmostrando 6
Characterization of cardiac involvement in children with LMNA-related muscular dystrophy.
Frontiers in cell and developmental biologyLamin-Related Congenital Muscular Dystrophy Alters Mechanical Signaling and Skeletal Muscle Growth.
International journal of molecular sciencesClinical spectrum and genetic variations of LMNA-related muscular dystrophies in a large cohort of Chinese patients.
Journal of medical geneticsCardiac diseases as a predictor warning of hereditary muscle diseases. The case of laminopathies.
Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of MyologyUp-regulation of Toll-like receptors 7 and 9 and its potential implications in the pathogenic mechanisms of LMNA-related myopathies.
Nucleus (Austin, Tex.)SMAD6 overexpression leads to accelerated myogenic differentiation of LMNA mutated cells.
Scientific reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Distrofia muscular congênita, tipo 1b.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Distrofia muscular congênita, tipo 1b
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Characterization of cardiac involvement in children with LMNA-related muscular dystrophy.
- Clinical spectrum and genetic variations of LMNA-related muscular dystrophies in a large cohort of Chinese patients.
- Lamin-Related Congenital Muscular Dystrophy Alters Mechanical Signaling and Skeletal Muscle Growth.
- Cardiac diseases as a predictor warning of hereditary muscle diseases. The case of laminopathies.Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology· 2019· PMID 31309180mais citado
- Up-regulation of Toll-like receptors 7 and 9 and its potential implications in the pathogenic mechanisms of LMNA-related myopathies.
- Biosynthesis and Regulation of Laminin-Binding O-Mannosyl Glycans Related to Muscular Dystrophy.
- Delayed presentation of muscular torticollis- an observational study.
- Laminin-α2 is required for the maintenance of the myotendinous junction in vivo.
- Clinico-genetic heterogeneity in Pakistani families affected with muscular dystrophies.
- Pearls & Oy-sters: Use of Short-Acting B-Agonist in DOK7-Related Congenital Myasthenic Syndrome Treatment.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:98893(Orphanet)
- OMIM OMIM:604801(OMIM)
- MONDO:0011486(MONDO)
- GARD:12586(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q32139634(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
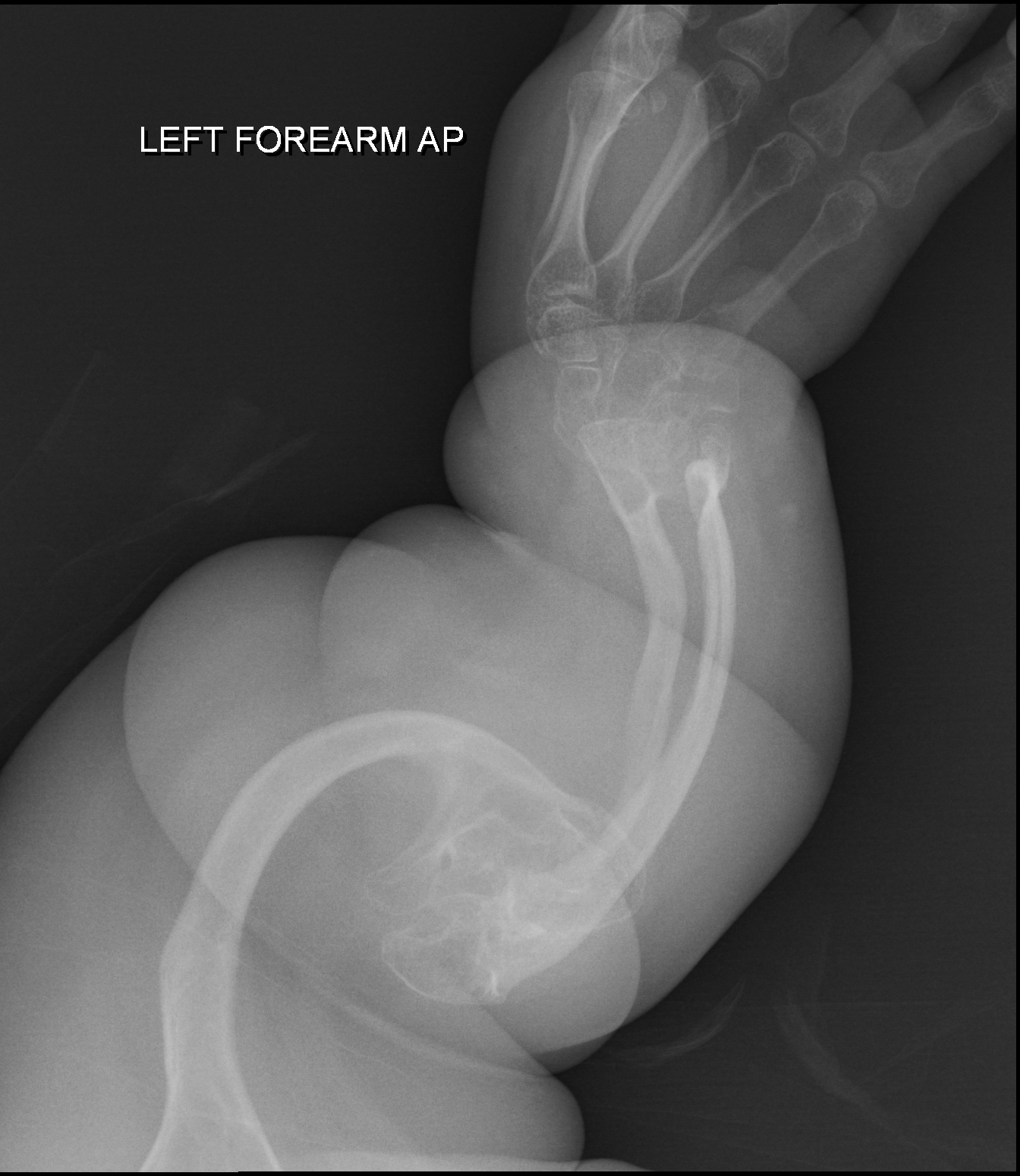
Distrofia muscular congênita, tipo 1b
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata