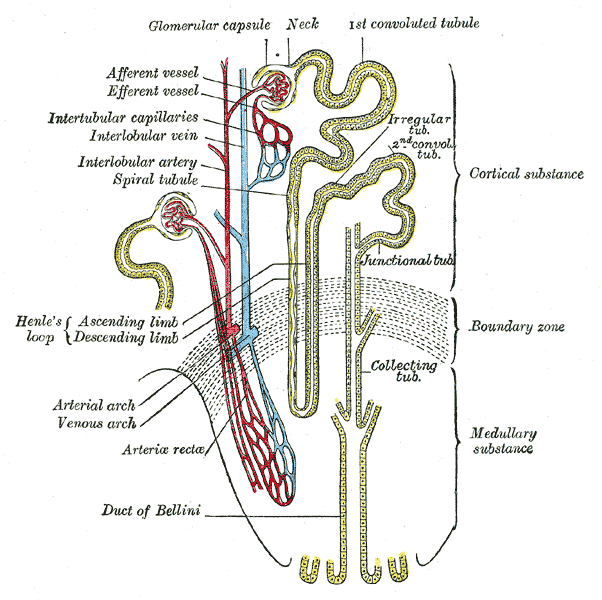
A doença Dent tipo 1 é um tipo de doença Dent com manifestações predominantemente renais.
Introdução
O que você precisa saber de cara
A doença Dent tipo 1 é um tipo de doença Dent com manifestações predominantemente renais.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 14 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 32 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: X-linked recessive.
Curadoria gene-doença
fontes oficiaisProton-coupled chloride transporter. Functions as antiport system and exchanges chloride ions against protons (PubMed:20466723). Important for normal acidification of the endosome lumen. May play an important role in renal tubular function. The CLC channel family contains both chloride channels and proton-coupled anion transporters that exchange chloride or another anion for protons. The absence of conserved gating glutamate residues is typical for family members that function as channels (Proba
Golgi apparatus membraneEndosome membraneCell membrane
Hypophosphatemic rickets, X-linked recessive
A renal disease belonging to the 'Dent disease complex', a group of disorders characterized by proximal renal tubular defect, hypercalciuria, nephrocalcinosis, and renal insufficiency. The spectrum of phenotypic features is remarkably similar in the various disorders, except for differences in the severity of bone deformities and renal impairment. XLHRR patients present with rickets or osteomalacia, hypophosphatemia due to decreased renal tubular phosphate reabsorption, hypercalciuria, and low molecular weight proteinuria. Patients develop nephrocalcinosis with progressive renal failure in adulthood. Female carriers may have asymptomatic hypercalciuria or hypophosphatemia only.
Variantes genéticas (ClinVar)
478 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 254 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença Dent tipo 1
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
4 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Molecular Mechanisms of CLCN5 Missense Mutations in Dent Disease Type 1: A Comprehensive Computational Analysis and Clinical Correlations in a Chinese Cohort.
Dent's disease, an X-linked recessive disorder predominantly affecting males, is characterized by nephrocalcinosis, nephrolithiasis, and a high risk of progression to end-stage renal disease. Dent's disease type 1, accounting for 60% of cases, caused by mutations in the CLCN5 gene encoding the chloride ion channel protein ClC-5, exhibits significant clinical heterogeneity and variability in disease progression. The lack of hotspot mutations poses challenges for genetic diagnosis and counselling, complicating the prediction of disease outcomes. This study systematically evaluated the functional and structural impacts of 181 CLCN5 missense mutations using computational tools, including PredictSNP, MAGPIE, and molecular dynamics simulations, to propose a robust method for improving genetic counselling and prognosis prediction. Our analysis identified mutations at the dimer interface and chloride selectivity filter as critical disruptors of ClC-5 function and stability. Notably, molecular dynamics simulations of L200R, P213L, and G512R mutations revealed that L200R significantly destabilized the protein structure. Clinical data from a multicentre cohort of Chinese patients with CLCN5 mutations corroborated our computational predictions, highlighting the essential role of helix O in ClC-5 function. By integrating bioinformatics analyses with clinical validation, this study provides molecular insights into Dent's disease heterogeneity and proposes a framework for enhancing genetic counselling and prognostic assessment for affected patients.
The Significance of FGF23 and 24,25-Dihydroxyvitamin D in Dent Disease Type 1.
Hypercalciuria is a prominent characteristic in Dent disease type 1 (DD1) and is associated with kidney stones and nephrocalcinosis. The objectives of this study were to assess fibroblast growth factor 23 (FGF23) and 24,25-dihydroxyvitamin D (24,25(OH)2D) in DD1 patients and investigate the effects of phosphate supplementation on urinary calcium excretion. Serum and 24-hour urine assessments from adult and pediatric DD1 patients (n=10 adults; n=9 pediatrics) were compared to adult control subjects with a history of idiopathic calcium kidney stones and hypercalciuria (n=9). Adult DD1 patients and control participants completed an oral phosphate supplementation intervention (1g/day x 14 days) with reassessment immediately following intervention. FGF23 was significantly lower in DD1 than in the control cohort (adults, p=0.006) and positively correlated with 24,25(OH)2D across all study cohorts. The concentrations of 24,25(OH)2D were low with conversion ratios (25-hydroxyvitamin D:24,25(OH)2D) exceeding the clinical reference limit for five of 10 adults and six of nine pediatric DD1 patients. The DD1 cohorts were then stratified by the 24,25(OH)2D ratio into "normal" and "low" 24,25(OH)2D. Adult DD1 patients with low 24,25(OH)2D (n=5) had lower FGF23, higher 1,25(OH)2D, greater urine calcium, and greater urine protein. Pediatric stratified data mirrored that in adults with the exception of no difference in serum 1,25(OH)2D. Phosphate supplementation was effective in decreasing urine calcium in both adult DD1 and control adult cohorts. Clinical measurement of 24,25(OH)2D is a novel and useful analysis for evaluating the severity of calcium and protein dysregulation in DD1. In addition, moderate phosphate supplementation effectively mitigates urine calcium excretion in DD1 adult patients.
Genetic and clinical phenotype of Dent disease in Chinese children and the etiological analysis of early - onset chronic kidney disease.
A prominent feature of Dent disease (DD) is the progressive decline in renal function, with 30% - 80% of male patients advancing to end-stage renal disease between the ages of 30 and 50 years. However, limited research exists on the chronic kidney disease (CKD) progression in pediatric patients with DD. This study aimed to retrospectively analyze the clinical features, genetic variant spectrum, and prognosis of pediatric patients with DD and explore the factors associated with early renal failure during childhood in these patients. We analyzed the genetic backgrounds, clinical phenotypes, and laboratory data of 23 unrelated patients with DD. All patients were males with low-molecular-weight proteinuria. CLCN5 variants were detected in 19 patients (Dent disease type 1, DD1), and OCRL variants were identified in 4 patients (Dent disease type 2, DD2). Sixteen mutations have not been reported previously. During follow-up, progression to CKD was documented in 7 patients: 6 with DD1 and 1 with DD2. CKD stages were distributed as follows: 4 patients at stage II, 2 at stage III, and 1 at stage V. Nephrolithiasis (100% vs 30.76%, P = 0.011), nephrocalcinosis (85.33% vs 15.38%, P = 0.010), and acute kideny injury (100% vs 0%, P < 0.001) were significantly more common in those CKD patients with DD1. This study expands the genetic spectrum of Dent disease and highlights that some pediatric patients may progress to CKD during childhood. CKD progression may be associated with early occurrences of nephrolithiasis, nephrocalcinosis, and acute kidney injury.
Phenotype and genotype analyses of 21 Chinese patients with Dent disease.
Dent disease is a rare X-linked recessively inherited renal tubulopathy, caused by variants in the CLCN5 (Dent disease type 1, DD1) and OCRL (Dent disease type 2, DD2) genes, and characterized by low molecular weight proteinuria, hypercalciuria, microscopic hematuria, or nephrocalcinosis. In the current study, we collected and analyzed clinical data and genetic testing results of 21 children diagnosed with Dent disease, who were hospitalized at the Department of Nephrology, Children's Hospital of Nanjing Medical University between January 2014 and August 2023, aiming to assist in the early diagnosis and treatment of these patients. Among the 21 patients, 16 (76.19%) had CLCN5 variants, and five (23.81%) had OCRL variants, and four of the variants were novel. All patients presented with low molecular weight proteinuria, 14 (66.67%) of whom had nephrotic-range proteinuria. Eight patients underwent renal biopsies because of diagnostic uncertainty. We transfected the novel CLCN5 missense variant (p.G222R) and OCRL missense variant (p.I371T) plasmids into both HEK293 and HK-2 cells and found a significantly lower expression of the OCRL1 protein in the transfected cells than in the wild-type cells ( P < 0.05). Moreover, we observed an extremely skewed X-chromosome inactivation pattern in a female patient carrying the same novel CLCN5 variant, as assessed by the human androgen receptor gene assay. These findings provide insight into the clinical characteristics of Dent disease in Chinese patients and may shed light on its pathogenesis.
Case Report: Early acute kidney failure in an 11-year-old boy with Dent disease type 1.
Dent disease type 1 (Dent 1) is a rare X-linked genetic condition which impacts kidney function and is caused by pathogenic variants in CLCN5. Affected males typically develop low molecular weight proteinuria, hypercalciuria, nephrocalcinosis, nephrolithiasis, and other symptoms. Kidney failure often occurs between the third to fifth decade of life. Here, we report an 11-year-old boy with Dent 1 and a severe kidney disease phenotype. The patient presented with flank pain, nocturnal enuresis, foamy urine, and increased urinary frequency. He was found to have nephrotic-range proteinuria, without hypoalbuminemia, and a significantly decreased estimated glomerular filtration rate at presentation. Further, he did not have hypercalciuria. His family history was remarkable for kidney disease among several relatives including a maternal half-brother and two sons of a maternal great aunt. Due to his symptoms and a strong family history, the patient underwent genetic testing that detected a novel pathogenic variant in CLCN5 [c.791dup (p.Ser265Glnfs*3)]. Given the variability of symptoms among family members and the early onset of severe symptoms in this young patient compared to prior literature, we encourage genetic testing for Dent disease in similarly affected individuals.
Publicações recentes
Molecular Mechanisms of CLCN5 Missense Mutations in Dent Disease Type 1: A Comprehensive Computational Analysis and Clinical Correlations in a Chinese Cohort.
The Significance of Fibroblast Growth Factor 23 and 24,25-Dihydroxyvitamin D in Dent Disease Type 1.
Genetic and clinical phenotype of Dent disease in Chinese children and the etiological analysis of early - onset chronic kidney disease.
Phenotype and genotype analyses of 21 Chinese patients with Dent disease.
Case Report: Early acute kidney failure in an 11-year-old boy with Dent disease type 1.
📚 EuropePMC136 artigos no totalmostrando 20
Molecular Mechanisms of CLCN5 Missense Mutations in Dent Disease Type 1: A Comprehensive Computational Analysis and Clinical Correlations in a Chinese Cohort.
Journal of cellular and molecular medicineThe Significance of FGF23 and 24,25-Dihydroxyvitamin D in Dent Disease Type 1.
Clinical journal of the American Society of Nephrology : CJASNGenetic and clinical phenotype of Dent disease in Chinese children and the etiological analysis of early - onset chronic kidney disease.
Italian journal of pediatricsPhenotype and genotype analyses of 21 Chinese patients with Dent disease.
Journal of biomedical researchCase Report: Early acute kidney failure in an 11-year-old boy with Dent disease type 1.
Frontiers in pediatricsA Focus on the Proximal Tubule Dysfunction in Dent Disease Type 1.
GenesGene therapy of Dent disease type 1 in newborn ClC-5 null mice for sustained transgene expression and gene therapy effects.
Gene therapy[Two cases of Dent disease type 1 with Bartter-like phenotype and literature review].
Zhonghua yi xue za zhiA female patient with Dent disease due to skewed X-chromosome inactivation.
Clinical kidney journalModeling Dent Disease Type 1 in Flies.
Kidney360The Apical Endocytic-Lysosomal Apparatus in CLCN5 Mutations with Phenotypic-Genotypic Correlations in Three Cases.
International journal of molecular sciencesDrosophila ClC-c Is a Homolog of Human CLC-5 and a New Model for Dent Disease Type 1.
Kidney360Hemizygous loss of function mutations in CLCN5 causing end-stage kidney disease without Dent disease phenotype.
Clinical kidney journalDent Disease Type 1: A Diagnostic Dilemma and Review.
CureusSmall molecules restore the function of mutant CLC5 associated with Dent disease.
Journal of cellular and molecular medicineA Novel CLCN5 Splice Site Mutation in a Boy with Incomplete Phenotype of Dent Disease.
Journal of pediatric geneticsMulticenter study of the clinical features and mutation gene spectrum of Chinese children with Dent disease.
Clinical geneticsDent disease: A window into calcium and phosphate transport.
Journal of cellular and molecular medicinePrevalence of low molecular weight proteinuria and Dent disease 1 CLCN5 mutations in proteinuric cohorts.
Pediatric nephrology (Berlin, Germany)Functional and transport analyses of CLCN5 genetic changes identified in Dent disease patients.
Physiological reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença Dent tipo 1.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença Dent tipo 1
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Molecular Mechanisms of CLCN5 Missense Mutations in Dent Disease Type 1: A Comprehensive Computational Analysis and Clinical Correlations in a Chinese Cohort.
- The Significance of FGF23 and 24,25-Dihydroxyvitamin D in Dent Disease Type 1.
- Genetic and clinical phenotype of Dent disease in Chinese children and the etiological analysis of early - onset chronic kidney disease.
- Phenotype and genotype analyses of 21 Chinese patients with Dent disease.
- Case Report: Early acute kidney failure in an 11-year-old boy with Dent disease type 1.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:93622(Orphanet)
- OMIM OMIM:300009(OMIM)
- MONDO:0010225(MONDO)
- GARD:1804(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55999527(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Doença Dent tipo 1
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata