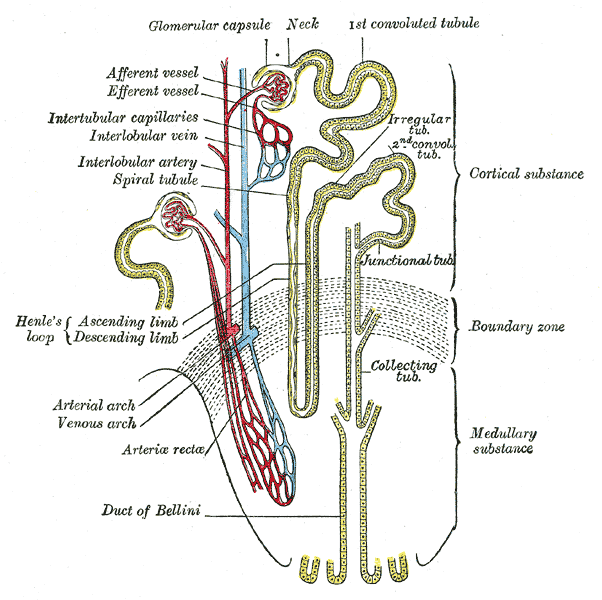
A doença de Dent é uma doença tubular renal genética rara, caracterizada por manifestações de disfunção do túbulo proximal.
Introdução
O que você precisa saber de cara
A doença de Dent é uma doença tubular renal genética rara, caracterizada por manifestações de disfunção do túbulo proximal.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 18 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 52 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: X-linked recessive.
Catalyzes the hydrolysis of the 5-position phosphate of phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) and phosphatidylinositol-3,4,5-bisphosphate (PtdIns(3,4,5)P3), with the greatest catalytic activity towards PtdIns(4,5)P2 (PubMed:10764818, PubMed:15474001, PubMed:7761412, PubMed:9430698). Able also to hydrolyze the 5-phosphate of inositol 1,4,5-trisphosphate and of inositol 1,3,4,5-tetrakisphosphate (PubMed:25869668, PubMed:7761412). Regulates traffic in the endosomal pathway by regula
Cytoplasmic vesicle, phagosome membraneEarly endosome membraneMembrane, clathrin-coated pitCell projection, cilium, photoreceptor outer segmentCell projection, ciliumCytoplasmic vesicleEndosomeGolgi apparatus, trans-Golgi networkLysosome
Lowe oculocerebrorenal syndrome
X-linked multisystem disorder affecting eyes, nervous system, and kidney. It is characterized by hydrophthalmia, cataract, intellectual disability, vitamin D-resistant rickets, aminoaciduria, and reduced ammonia production by the kidney. Ocular abnormalities include cataract, glaucoma, microphthalmos, and decreased visual acuity. Developmental delay, hypotonia, behavior abnormalities, and areflexia are also present. Renal tubular involvement is characterized by impaired reabsorption of bicarbonate, amino acids, and phosphate. Musculoskeletal abnormalities such as joint hypermobility, dislocated hips, and fractures may develop as consequences of renal tubular acidosis and hypophosphatemia. Cataract is the only significant manifestation in carriers and is detected by slit-lamp examination.
Proton-coupled chloride transporter. Functions as antiport system and exchanges chloride ions against protons (PubMed:20466723). Important for normal acidification of the endosome lumen. May play an important role in renal tubular function. The CLC channel family contains both chloride channels and proton-coupled anion transporters that exchange chloride or another anion for protons. The absence of conserved gating glutamate residues is typical for family members that function as channels (Proba
Golgi apparatus membraneEndosome membraneCell membrane
Hypophosphatemic rickets, X-linked recessive
A renal disease belonging to the 'Dent disease complex', a group of disorders characterized by proximal renal tubular defect, hypercalciuria, nephrocalcinosis, and renal insufficiency. The spectrum of phenotypic features is remarkably similar in the various disorders, except for differences in the severity of bone deformities and renal impairment. XLHRR patients present with rickets or osteomalacia, hypophosphatemia due to decreased renal tubular phosphate reabsorption, hypercalciuria, and low molecular weight proteinuria. Patients develop nephrocalcinosis with progressive renal failure in adulthood. Female carriers may have asymptomatic hypercalciuria or hypophosphatemia only.
Variantes genéticas (ClinVar)
925 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 357 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
9 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença de Dent
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
5 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
12 ensaios clínicos encontrados, 5 ativos.
Publicações mais relevantes
Molecular Mechanisms of CLCN5 Missense Mutations in Dent Disease Type 1: A Comprehensive Computational Analysis and Clinical Correlations in a Chinese Cohort.
Dent's disease, an X-linked recessive disorder predominantly affecting males, is characterized by nephrocalcinosis, nephrolithiasis, and a high risk of progression to end-stage renal disease. Dent's disease type 1, accounting for 60% of cases, caused by mutations in the CLCN5 gene encoding the chloride ion channel protein ClC-5, exhibits significant clinical heterogeneity and variability in disease progression. The lack of hotspot mutations poses challenges for genetic diagnosis and counselling, complicating the prediction of disease outcomes. This study systematically evaluated the functional and structural impacts of 181 CLCN5 missense mutations using computational tools, including PredictSNP, MAGPIE, and molecular dynamics simulations, to propose a robust method for improving genetic counselling and prognosis prediction. Our analysis identified mutations at the dimer interface and chloride selectivity filter as critical disruptors of ClC-5 function and stability. Notably, molecular dynamics simulations of L200R, P213L, and G512R mutations revealed that L200R significantly destabilized the protein structure. Clinical data from a multicentre cohort of Chinese patients with CLCN5 mutations corroborated our computational predictions, highlighting the essential role of helix O in ClC-5 function. By integrating bioinformatics analyses with clinical validation, this study provides molecular insights into Dent's disease heterogeneity and proposes a framework for enhancing genetic counselling and prognostic assessment for affected patients.
The Significance of FGF23 and 24,25-Dihydroxyvitamin D in Dent Disease Type 1.
Hypercalciuria is a prominent characteristic in Dent disease type 1 (DD1) and is associated with kidney stones and nephrocalcinosis. The objectives of this study were to assess fibroblast growth factor 23 (FGF23) and 24,25-dihydroxyvitamin D (24,25(OH)2D) in DD1 patients and investigate the effects of phosphate supplementation on urinary calcium excretion. Serum and 24-hour urine assessments from adult and pediatric DD1 patients (n=10 adults; n=9 pediatrics) were compared to adult control subjects with a history of idiopathic calcium kidney stones and hypercalciuria (n=9). Adult DD1 patients and control participants completed an oral phosphate supplementation intervention (1g/day x 14 days) with reassessment immediately following intervention. FGF23 was significantly lower in DD1 than in the control cohort (adults, p=0.006) and positively correlated with 24,25(OH)2D across all study cohorts. The concentrations of 24,25(OH)2D were low with conversion ratios (25-hydroxyvitamin D:24,25(OH)2D) exceeding the clinical reference limit for five of 10 adults and six of nine pediatric DD1 patients. The DD1 cohorts were then stratified by the 24,25(OH)2D ratio into "normal" and "low" 24,25(OH)2D. Adult DD1 patients with low 24,25(OH)2D (n=5) had lower FGF23, higher 1,25(OH)2D, greater urine calcium, and greater urine protein. Pediatric stratified data mirrored that in adults with the exception of no difference in serum 1,25(OH)2D. Phosphate supplementation was effective in decreasing urine calcium in both adult DD1 and control adult cohorts. Clinical measurement of 24,25(OH)2D is a novel and useful analysis for evaluating the severity of calcium and protein dysregulation in DD1. In addition, moderate phosphate supplementation effectively mitigates urine calcium excretion in DD1 adult patients.
Ten tips on the work-up and management of CKD patients with nephrolithiasis.
Historically, the management of patients with nephrolithiasis has been primarily delivered by urologists. In recent decades nephrologists have had an increasing role in caring for these patients not only in advanced stages of chronic kidney disease (CKD) but also in the diagnosis and management of underlying systemic disease, and metabolic and genetic risk factors. Globally the burden of kidney stone disease has been increasing, with metabolic disorders strongly associated with nephrolithiasis and stone-promoting urinary risk factors, including hypercalciuria, hyperoxaluria and hypocitraturia. At a healthcare level, kidney stones account for a relatively large number of emergency department visits, while individually the impact of kidney stones and the fear of recurrence often result in loss of work and high levels of psychological distress, in addition to an increased CKD risk. Kidney stones in the context of CKD of unknown cause can be the signature of an inherited disorder such as Dent disease, primary hyperoxaluria or adenine phosphoribosyltransferase deficiency (among others). Diagnosing these diseases is imperative in the current age of new treatments (e.g. small interfering RNA therapies), to avoid further deterioration of kidney function and disease recurrence post-transplantation. We outline 10 practical tips to guide clinicians in the diagnostic evaluation and management of nephrolithiasis in CKD.
Dual-Genetic Etiology in an Atypical Dent Disease Phenotype Which Combines Features of Focal Segmental Glomerulosclerosis and Ellis-Van Creveld-Like Syndrome: A Case Report.
Dent disease (DD) is an X-linked recessive renal disorder characterized by features of incomplete Fanconi syndrome. DD varies in clinical presentation, manifesting with proteinuria alone or in combination with nephrocalcinosis/nephrolithiasis, and with or without chronic kidney disease, posing a challenge to clinical diagnosis. The genetic basis of DD is not completely known; about 25-35% of DD cases lack mutations in the disease-causing CLCN5 and OCRL genes. This case report represents a rare example of a patient initially suspected of having DD, but through whole exome sequencing (WES) was found to harbor pathogenic variants in the WT1 and EVC2 genes, suggesting a dual-genetic etiology mimicking DD. We describe a young man with a renal phenotype resembling DD associated with nephrotic syndrome, focal segmental glomerulosclerosis (FSGS), tubular microcysts, and a significant family history of kidney disease. Also present was an extrarenal phenotype with short stature, narrow chest, recurrent upper respiratory tract infections, teeth anomalies and hypertension. We identified in the WT1 gene the heterozygous ultrarare missense variant (NM_024426.6:c.1088C>T p.Thr363Met), classified as a variant of uncertain significance, and in the EVC2 gene the heterozygous nonsense variant (NM_147127.5:c.2833C>T p.Arg945Ter), classified as pathogenic. The clinical phenotype combines WT1-related FSGS with a rare tubular phenotype of Ellis-van Creveld-like syndrome (EVC). This case report provides insights into the phenotypic complexity of hereditary nephropathies and the diagnostic challenge posed by overlapping glomerular and tubular presentations. WES enabled us to expand our knowledge of the genetics of kidney diseases in adults and to reclassify the patient's nephropathy.
Empagliflozin does not prevent progression of Dent's disease type 1 in a mouse model.
Dent's disease is a rare inherited renal disorder characterized by generalized proximal tubule dysfunction with low molecular weight proteinuria, hypercalciuria, and urinary loss of other solutes. The disease is progressive and leads to chronic kidney disease. To study the mechanisms involved in its progression, we generated a knock-in mouse model displaying a classical Dent's disease type 1 phenotype. Currently, no targeted therapy exists for Dent's disease; treatment strategies primarily aim to slow the progression of specific clinical aspects. Accordingly, empagliflozin [a sodium-glucose cotransporter 2 (SGLT2) inhibitor] known to exert nephroprotective effects and to slow down the decrease of the glomerular filtration rate in diabetic and non-diabetic patients with chronic kidney disease, was administered to the knock-in mice. We demonstrated that empagliflozin administration reduces renal and urinary levels of the marker of tubular damage, Lipocalin-2 (LCN2). However, we observed that this preventive treatment does not alleviate low molecular weight proteinuria, hypercalciuria, inflammation, renal fibrosis or the decline of the glomerular filtration rate. Overall, our findings suggest that SGLT2 inhibition with empagliflozin does not prevent the progression of Dent's disease type 1 towards chronic kidney disease.
Publicações recentes
Dent Disease 1 Associated with a Rare Novel Renal Chloride Channel 5 Variant in a Chinese Family.
Molecular Mechanisms of CLCN5 Missense Mutations in Dent Disease Type 1: A Comprehensive Computational Analysis and Clinical Correlations in a Chinese Cohort.
Coexisting genetic kidney disease explains many cases of 'familial' IgA nephropathy where the proband has biopsy-confirmed mesangial IgA deposits.
The Significance of Fibroblast Growth Factor 23 and 24,25-Dihydroxyvitamin D in Dent Disease Type 1.
Ten tips on the work-up and management of CKD patients with nephrolithiasis.
📚 EuropePMC136 artigos no totalmostrando 183
Molecular Mechanisms of CLCN5 Missense Mutations in Dent Disease Type 1: A Comprehensive Computational Analysis and Clinical Correlations in a Chinese Cohort.
Journal of cellular and molecular medicineCoexisting genetic kidney disease explains many cases of 'familial' IgA nephropathy where the proband has biopsy-confirmed mesangial IgA deposits.
Frontiers in medicineThe Significance of FGF23 and 24,25-Dihydroxyvitamin D in Dent Disease Type 1.
Clinical journal of the American Society of Nephrology : CJASNTen tips on the work-up and management of CKD patients with nephrolithiasis.
Clinical kidney journalDual-Genetic Etiology in an Atypical Dent Disease Phenotype Which Combines Features of Focal Segmental Glomerulosclerosis and Ellis-Van Creveld-Like Syndrome: A Case Report.
Case reports in nephrology and dialysisGenetic and clinical phenotype of Dent disease in Chinese children and the etiological analysis of early - onset chronic kidney disease.
Italian journal of pediatricsClinical variation in Lowe syndrome: what and how?
Frontiers in cell and developmental biologyThree intronic variants altering RNA splicing were identified in the CLCN5 gene by minigene assay.
BMC medical genomicsLong-term recombinant human growth hormone therapy in Dent's disease type 1.
Endokrynologia PolskaBioinformatics analysis of a CLCN5 geneframeshift mutation in a patient with Dent disease.
Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciencesDent Disease 1 Presented Early with Bartter-Like Syndrome Features and Rickets: A Case Report.
Case reports in nephrology and dialysisEmpagliflozin does not prevent progression of Dent's disease type 1 in a mouse model.
Experimental physiologyThe inositol 5-phosphatases OCRL and INPP5B: Cellular functions and roles in disease.
Biochimica et biophysica acta. Molecular and cell biology of lipids[Clinical and genetic analysis of a patient with Dent disease due to hemizygous variant of the CLCN5 gene].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsComprehensive Splice Pattern Analysis for Previously Reported OCRL Splicing Variants and Their Phenotypic Contributions.
Kidney international reportsPhenotypes and the Importance of Genetic Analysis in Adult Patients with Nephrolithiasis and/or Nephrocalcinosis: A Single-Center Experience.
GenesPhenotype and genotype analyses of 21 Chinese patients with Dent disease.
Journal of biomedical researchUnderstanding Rare Kidney Stone Diseases: A Review.
American journal of kidney diseases : the official journal of the National Kidney FoundationTubular proteinuria due to hereditary endocytic receptor disorder of the proximal tubule: Dent disease and chronic benign proteinuria.
Pediatric nephrology (Berlin, Germany)Clinical features and genetic analysis of nine Chinese children with Dent disease and identification of three novel CLCN5 and OCRL variants.
Renal failureDent disease: clinical practice recommendations.
Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal AssociationCase Report: Early acute kidney failure in an 11-year-old boy with Dent disease type 1.
Frontiers in pediatricsDent's disease: case series from a single center.
The Turkish journal of pediatricsA narrative review of monogenic disorders causing nephrolithiasis and chronic kidney disease.
Nephrology (Carlton, Vic.)A Focus on the Proximal Tubule Dysfunction in Dent Disease Type 1.
GenesPediatric Dent disease presenting with rickets and end-stage renal disease: case report and literature review.
The Journal of international medical researchGene therapy of Dent disease type 1 in newborn ClC-5 null mice for sustained transgene expression and gene therapy effects.
Gene therapyPrevalence of kidney failure in adults diagnosed with hereditary tubulopathies.
Journal of nephrology[Two cases of Dent disease type 1 with Bartter-like phenotype and literature review].
Zhonghua yi xue za zhi4-Phenylbutyric Acid Treatment Reduces Low-Molecular-Weight Proteinuria in a Clcn5 Knock-in Mouse Model for Dent Disease-1.
International journal of molecular sciencesA novel transgenic mouse model highlights molecular disruptions involved in the pathogenesis of Dent disease 1.
GeneA female patient with Dent disease due to skewed X-chromosome inactivation.
Clinical kidney journalModeling Dent Disease Type 1 in Flies.
Kidney360Clinical features and genetic analysis of 15 Chinese children with dent disease.
Renal failureRenal antiporter ClC-5 regulates collagen I/IV through the β-catenin pathway and lysosomal degradation.
Life science allianceIsolation and characterization of exosome-enriched urinary extracellular vesicles from Dent's disease type 1 Spanish patients.
NefrologiaPrenatal diagnosis of dent disease type I with a nonsense pathogenic variant in CLCN5: a case study.
BMC medical genomicsHistologic and Clinical Factors Associated with Kidney Outcomes in IgA Vasculitis Nephritis.
Clinical journal of the American Society of Nephrology : CJASNThe Apical Endocytic-Lysosomal Apparatus in CLCN5 Mutations with Phenotypic-Genotypic Correlations in Three Cases.
International journal of molecular sciencesDrosophila ClC-c Is a Homolog of Human CLC-5 and a New Model for Dent Disease Type 1.
Kidney360A Case of Hidradenitis Suppurativa in a Genetically Confirmed Lowe Syndrome Patient.
Annals of dermatologyDent disease 1-linked novel CLCN5 mutations result in aberrant location and reduced ion currents.
International journal of biological macromoleculesA missense mutant of ocrl1 promotes apoptosis of tubular epithelial cells and disrupts endocytosis and the cell cycle of podocytes in Dent-2 Disease.
Cell communication and signaling : CCSCharacterization of pre-mRNA Splicing Defects Caused by CLCN5 and OCRL Mutations and Identification of Novel Variants Associated with Dent Disease.
BiomedicinesA novel likely pathogenic CLCN5 variant in Dent's disease.
BMC nephrologyGenotypic variability in patients with clinical diagnosis of Bartter syndrome type 3.
Scientific reportsPseudo-Bartter syndrome in an infant without obvious underlying conditions: A case report.
Clinical pediatric endocrinology : case reports and clinical investigations : official journal of the Japanese Society for Pediatric EndocrinologyGeneration of a human induced pluripotent stem cell line from a patient with dent disease.
Stem cell researchThe Site and Type of CLCN5 Genetic Variation Impact the Resulting Dent Disease-1 Phenotype.
Kidney international reportsResearch progress on renal calculus associate with inborn error of metabolism.
Zhejiang da xue xue bao. Yi xue ban = Journal of Zhejiang University. Medical sciences[A rare tubulopathy: Dent's disease in the background of focal segmental glomerular sclerosis].
Orvosi hetilapAnalysis of the ratio of urinary beta-2-microglobulin to total protein concentration in children with isolated tubulointerstitial disease.
Clinical and experimental nephrologyDent disease manifesting as nephrotic syndrome.
Intractable & rare diseases researchImpaired Endosome Maturation Mediates Tubular Proteinuria in Dent Disease Cell Culture and Mouse Models.
Journal of the American Society of Nephrology : JASNHemizygous loss of function mutations in CLCN5 causing end-stage kidney disease without Dent disease phenotype.
Clinical kidney journal[Short-term efficacy of dapagliflozin in children with hereditary proteinuric kidney disease].
Zhonghua er ke za zhi = Chinese journal of pediatricsEmerging Perspectives on the Rare Tubulopathy Dent Disease: Is Glomerular Damage a Direct Consequence of ClC-5 Dysfunction?
International journal of molecular sciencesA Study on the CLCN5 Gene in Iranian Patients: A Report of Novel and Recurrent Mutations.
NephronPrenatal ultrasound findings of X-linked congenital cataracts: case report and description of a novel variant.
American journal of translational researchDent disease presenting with nyctalopia and electroretinographic correlates of vitamin A deficiency.
American journal of ophthalmology case reportsA novel CLCN5 frame shift mutation responsible for Dent disease 1: Case report.
Frontiers in pediatricsClinical and genetic characteristics of Dent's disease type 1 in Europe.
Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal AssociationLentiviral vector mediated gene therapy for type I Dent disease ameliorates Dent disease-like phenotypes for three months in ClC-5 null mice.
Molecular therapy. Methods & clinical developmentBe aware of underlying Dent disease in young boys with massive proteinuria.
Pediatrics international : official journal of the Japan Pediatric Society[Current approach to management of staghorn nephrolithiasis. Literature review. Part 2].
Urologiia (Moscow, Russia : 1999)Young Adults With Hereditary Tubular Diseases: Practical Aspects for Adult-Focused Colleagues.
Advances in chronic kidney diseaseMegalin, a multi-ligand endocytic receptor, and its participation in renal function and diseases: A review.
Life sciencesGenetic and clinical profile of patients with hypophosphatemic rickets.
European journal of medical geneticsX-Linked Kidney Disorders in Women.
Seminars in nephrologyGenetic testing enables a precision medicine approach for nephrolithiasis and nephrocalcinosis in pediatrics: a single-center cohort.
Molecular genetics and genomics : MGGDent-2 disease with a Bartter-like phenotype caused by the Asp631Glu mutation in the OCRL gene.
BMC nephrologyDent Disease Type 1: A Diagnostic Dilemma and Review.
CureusMolecular Diagnosis of Primary Hyperoxaluria Type 1 and Distal Renal Tubular Acidosis in Moroccan Patients With Nephrolithiasis and/or Nephrocalcinosis.
CureusRickets guidance: part II-management.
Pediatric nephrology (Berlin, Germany)Renal Expression of CLC-5 and Megalin/Cubilin in Dent-1 Disease With Nonsense Mutations of CLCN5 Gene.
Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology SocietyA case of Dent disease type 2 with large deletion of OCRL diagnosed after close examination of a school urinary test.
CEN case reportsComprehensive Genetic Analysis Reveals Complexity of Monogenic Urinary Stone Disease.
Kidney international reportsGenotype Phenotype Correlation in Dent Disease 2 and Review of the Literature: OCRL Gene Pleiotropism or Extreme Phenotypic Variability of Lowe Syndrome?
GenesBartter-Like Syndrome as the Initial Presentation of Dent Disease 1: A Case Report.
Frontiers in pediatricsIdentification of novel OCRL isoforms associated with phenotypic differences between Dent disease-2 and Lowe syndrome.
Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal AssociationEstablishment of an induced pluripotent stem cell line (NCKDi003-A) from a patient with X-linked Dent disease (X-Dent) carrying the hemizygote mutation p. T277P (c. 829A > C) in the CLCN5 gene.
Stem cell researchAuto-inhibitory intramolecular S5/S6 interaction in the TRPV6 channel regulates breast cancer cell migration and invasion.
Communications biologyUrinary FABP1 is a biomarker for impaired proximal tubular protein reabsorption and is synergistically enhanced by concurrent liver injury.
The Journal of pathologyA 3D Renal Proximal Tubule on Chip Model Phenocopies Lowe Syndrome and Dent II Disease Tubulopathy.
International journal of molecular sciencesNovel Dent disease 1 cellular models reveal biological processes underlying ClC-5 loss-of-function.
Human molecular geneticsGlomerular podocyte dysfunction in inherited renal tubular disease.
World journal of pediatrics : WJPDiversity of functional alterations of the ClC-5 exchanger in the region of the proton glutamate in patients with Dent disease 1.
Human mutationDifferential diagnosis of perinatal Bartter, Bartter and Gitelman syndromes.
Clinical kidney journalEstablishment of an induced pluripotent stem cell line (WMUi016-A) from a patient with X-linked Dent disease (X-Dent) carrying the hemizygote mutation p.R718* (c.2152C > T) in the CLCN5 gene.
Stem cell research[Nephrocalcinosis in children].
Nephrologie & therapeutiqueClinical manifestation and genetic findings in three boys with low molecular Weight Proteinuria - three case reports for exploring Dent Disease and Fanconi syndrome.
BMC nephrologyAtypical presentation of Dent disease in a patient with interstitial Xp11.22 deletion.
Journal of nephrologyNovel Fanconi renotubular syndromes provide insights in proximal tubule pathophysiology.
American journal of physiology. Renal physiologySmall molecules restore the function of mutant CLC5 associated with Dent disease.
Journal of cellular and molecular medicineDent Disease Type 2 as a Cause of Focal Segmental Glomerulosclerosis in a 6-Year-Old Boy: A Case Report.
Frontiers in pediatricsA young man with recurrent kidney stones and renal failure.
Clinical nephrology. Case studiesFrom Skin to Kidneys: Cutaneous Clues of Renal Disease in Children.
Dermatology practical & conceptualMaking a Dent in Dent Disease.
Function (Oxford, England)The phosphoinositide 3-kinase inhibitor alpelisib restores actin organization and improves proximal tubule dysfunction in vitro and in a mouse model of Lowe syndrome and Dent disease.
Kidney internationalGenetics and phenotypic heterogeneity of Dent disease: the dark side of the moon.
Human geneticsNonO Is a Novel Co-factor of PRDM1 and Regulates Inflammatory Response in Monocyte Derived-Dendritic Cells.
Frontiers in immunologyGenetic and pathological findings in a boy with psoriasis and C3 glomerulonephritis: A case report and literature review.
Molecular genetics & genomic medicineComparison of clinical and genetic characteristics between Dent disease 1 and Dent disease 2.
Pediatric nephrology (Berlin, Germany)Onset mechanism of a female patient with Dent disease 2.
Clinical and experimental nephrologyA rare case of nephrotic syndrome associated with Dent's disease: a case report.
CEN case reportsPhenotypic spectrum and antialbuminuric response to angiotensin converting enzyme inhibitor and angiotensin receptor blocker therapy in pediatric Dent disease.
Molecular genetics & genomic medicineIncomplete cryptic splicing by an intronic mutation of OCRL in patients with partial phenotypes of Lowe syndrome.
Journal of human geneticsCase report: a Chinese girl with dent disease 1 and turner syndrome due to a hemizygous CLCN5 gene mutation and Isochromosome (Xq).
BMC nephrologyTranscriptome analysis of neural progenitor cells derived from Lowe syndrome induced pluripotent stem cells: identification of candidate genes for the neurodevelopmental and eye manifestations.
Journal of neurodevelopmental disordersFrom protein uptake to Dent disease: An overview of the CLCN5 gene.
GeneDent disease: classification, heterogeneity and diagnosis.
World journal of pediatrics : WJPA case of Type 1 Dent disease presenting with isolated persistent proteinuria.
Turk pediatri arsiviEtiological Profile of Nephrocalcinosis in Children from Southern India.
Indian pediatricsFunctional analysis of suspected splicing variants in CLCN5 gene in Dent disease 1.
Clinical and experimental nephrologyPolyclonal gammopathy in an adolescent affected by Dent disease 2 and hidradenitis suppurativa.
International journal of dermatologyGenetic Analyses in Dent Disease and Characterization of CLCN5 Mutations in Kidney Biopsies.
International journal of molecular sciencesNephrocalcinosis: A Review of Monogenic Causes and Insights They Provide into This Heterogeneous Condition.
International journal of molecular sciencesCl- and H+ coupling properties and subcellular localizations of wildtype and disease-associated variants of the voltage-gated Cl-/H+ exchanger ClC-5.
The Journal of biological chemistryClinical and genetic analysis of Dent disease with nephrotic range albuminuria in Shaanxi, China.
Science China. Life sciencesA Novel CLCN5 Splice Site Mutation in a Boy with Incomplete Phenotype of Dent Disease.
Journal of pediatric geneticsLowe syndrome identified in the offspring of an oocyte donor who was an unknown carrier of a de novo mutation: a case report and review of the literature.
Journal of medical case reportsMulticenter study of the clinical features and mutation gene spectrum of Chinese children with Dent disease.
Clinical geneticsLiving Kidney Donation in a Type 1 Dent's Disease Patient from His Mother.
Kidney & blood pressure researchDent disease: A window into calcium and phosphate transport.
Journal of cellular and molecular medicineUrinary apolipoprotein AI in children with kidney disease.
Pediatric nephrology (Berlin, Germany)Prevalence of low molecular weight proteinuria and Dent disease 1 CLCN5 mutations in proteinuric cohorts.
Pediatric nephrology (Berlin, Germany)C-Terminal Fibroblast Growth Factor-23 Levels in Non-Nutritional Hypophosphatemic Rickets.
Indian journal of pediatricsFamilial Xp11.22 microdeletion including SHROOM4 and CLCN5 is associated with intellectual disability, short stature, microcephaly and Dent disease: a case report.
BMC medical genomicsOCRL deficiency impairs endolysosomal function in a humanized mouse model for Lowe syndrome and Dent disease.
Human molecular geneticsNext-Generation Sequencing in Early Diagnosis of Dent Disease 1: Two Case Reports.
Frontiers in medicineBarttin Regulates the Subcellular Localization and Posttranslational Modification of Human Cl-/H+ Antiporter ClC-5.
Frontiers in physiologyPhosphoinositides in the kidney.
Journal of lipid researchModeling the neuropsychiatric manifestations of Lowe syndrome using induced pluripotent stem cells: defective F-actin polymerization and WAVE-1 expression in neuronal cells.
Molecular autismClinical Approach to Proximal Renal Tubular Acidosis in Children.
Advances in chronic kidney diseaseSevere Osteomalacia with Dent Disease Caused by a Novel Intronic Mutation of the CLCN5 gene.
Internal medicine (Tokyo, Japan)Congenital and acquired diseases related to stone formation.
Current opinion in urologyCongenital chloride diarrhea needs to be distinguished from Bartter and Gitelman syndrome.
Journal of human geneticsA novel CLCN5 pathogenic mutation supports Dent disease with normal endosomal acidification.
Human mutationIncidental Detection of Dent-2 Disease in an Infant with Febrile Proteinuria.
Medical principles and practice : international journal of the Kuwait University, Health Science CentreA novel mutation of Dent's disease in an 11-year-old male with nephrolithiasis and nephrocalcinosis.
Archivos argentinos de pediatria[Clinical features and genetic variants of Dent disease in 10 children].
Zhonghua er ke za zhi = Chinese journal of pediatricsThe ratio of urinary α1-microglobulin to microalbumin can be used as a diagnostic criterion for tubuloproteinuria.
Intractable & rare diseases researchHypercalciuria and nephrolithiasis: Expanding the renal phenotype of Donnai-Barrow syndrome.
Clinical geneticsDevelopment of ultra-deep targeted RNA sequencing for analyzing X-chromosome inactivation in female Dent disease.
Journal of human geneticsPatients affected by dent disease 2 could be predisposed to hidradenitis suppurativa.
Journal of the European Academy of Dermatology and Venereology : JEADVIdentification of co-occurrence in a patient with Dent's disease and ADA2-deficiency by exome sequencing.
GeneEarly Recognition and Management of Rare Kidney Stone Disorders.
Urologic nursingThe first Sri Lankan family with Dent disease-1 due to a pathogenic variant in the CLCN5 gene: a case report.
BMC research notesGenetic Analysis of Dent's Disease and Functional Research of CLCN5 Mutations.
DNA and cell biologyDent disease in Poland: what we have learned so far?
International urology and nephrologyThe 5-phosphatase OCRL in Lowe syndrome and Dent disease 2.
Nature reviews. NephrologyDent disease: Same CLCN5 mutation but different phenotypes in two brothers in China.
Intractable & rare diseases researchDiagnosis and treatment of Dent disease in 10 Chinese boys.
Intractable & rare diseases researchNanotubes, the fast track to treatment of Dent disease?
Kidney internationalBone marrow transplantation improves proximal tubule dysfunction in a mouse model of Dent disease.
Kidney internationalDigenic mutations of human OCRL paralogs in Dent's disease type 2 associated with Chiari I malformation.
Human genome variationDent's disease complicated by nephrotic syndrome: A case report.
Intractable & rare diseases researchKidney Tubular Ablation of Ocrl/Inpp5b Phenocopies Lowe Syndrome Tubulopathy.
Journal of the American Society of Nephrology : JASNPhenotype of Dent Disease in a Cohort of Indian Children.
Indian pediatricsReceptor-Mediated Endocytosis in the Proximal Tubule.
Annual review of physiologyAre filtered plasma proteins processed in the same way by the kidney?
Journal of theoretical biologyDecreased urinary excretion of the ectodomain form of megalin (A-megalin) in children with OCRL gene mutations.
Pediatric nephrology (Berlin, Germany)Proteinuria in Dent disease: a review of the literature.
Pediatric nephrology (Berlin, Germany)Phenotypic variability of Dent disease in a large New Zealand kindred.
Pediatric nephrology (Berlin, Germany)Glomerular Pathology in Dent Disease and Its Association with Kidney Function.
Clinical journal of the American Society of Nephrology : CJASNObservations of a large Dent disease cohort.
Kidney internationalDoes Dent disease remain an underrecognized cause for young boys with focal glomerulosclerosis?
Pediatrics international : official journal of the Japan Pediatric SocietyDent Disease in Chinese Children and Findings from Heterozygous Mothers: Phenotypic Heterogeneity, Fetal Growth, and 10 Novel Mutations.
The Journal of pediatricsFunctional and transport analyses of CLCN5 genetic changes identified in Dent disease patients.
Physiological reportsA pure chloride channel mutant of CLC-5 causes Dent's disease via insufficient V-ATPase activation.
Pflugers Archiv : European journal of physiologyThe oculocerebrorenal syndrome of Lowe: an update.
Pediatric nephrology (Berlin, Germany)On the Origin of Urinary Renin: A Translational Approach.
Hypertension (Dallas, Tex. : 1979)[Renal hypophosphatemia:pathophysiology and treatment].
Clinical calcium[Clinical and genetic analysis of Dent disease in 4 Chinese children].
Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatricsNephrolithiasis and Nephrocalcinosis in Children - Metabolic and Genetic Factors.
Pediatric endocrinology reviews : PEROCRL1 engages with the F-BAR protein pacsin 2 to promote biogenesis of membrane-trafficking intermediates.
Molecular biology of the cellNephrolithiasis, kidney failure and bone disorders in Dent disease patients with and without CLCN5 mutations.
SpringerPlusUrine proteome analysis in Dent's disease shows high selective changes potentially involved in chronic renal damage.
Journal of proteomicsDent disease in children: diagnostic and therapeutic considerations.
Clinical nephrologyNephrotic-range Albuminuria as the presenting symptom of Dent-2 disease.
Italian journal of pediatricsMutation Update of the CLCN5 Gene Responsible for Dent Disease 1.
Human mutationCharacterization of 26 deletion CNVs reveals the frequent occurrence of micro-mutations within the breakpoint-flanking regions and frequent repair of double-strand breaks by templated insertions derived from remote genomic regions.
Human geneticsDouble Xp11.22 deletion including SHROOM4 and CLCN5 associated with severe psychomotor retardation and Dent disease.
Molecular cytogeneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença de Dent.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença de Dent
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Molecular Mechanisms of CLCN5 Missense Mutations in Dent Disease Type 1: A Comprehensive Computational Analysis and Clinical Correlations in a Chinese Cohort.
- The Significance of FGF23 and 24,25-Dihydroxyvitamin D in Dent Disease Type 1.
- Ten tips on the work-up and management of CKD patients with nephrolithiasis.
- Dual-Genetic Etiology in an Atypical Dent Disease Phenotype Which Combines Features of Focal Segmental Glomerulosclerosis and Ellis-Van Creveld-Like Syndrome: A Case Report.
- Empagliflozin does not prevent progression of Dent's disease type 1 in a mouse model.
- Dent Disease 1 Associated with a Rare Novel Renal Chloride Channel 5 Variant in a Chinese Family.
- Coexisting genetic kidney disease explains many cases of 'familial' IgA nephropathy where the proband has biopsy-confirmed mesangial IgA deposits.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1652(Orphanet)
- MONDO:0015612(MONDO)
- GARD:13105(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q55999527(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Doença de Dent
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov