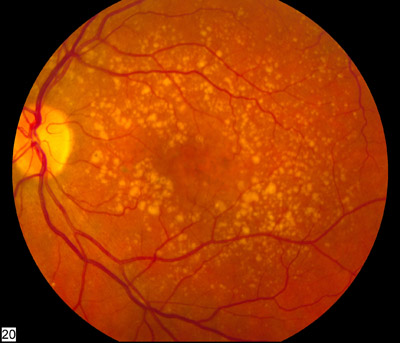
A distrofia retiniana em favo de mel de Doyne (DHRD) é uma condição que afeta os olhos e causa perda de visão. É caracterizada por pequenas manchas brancas, redondas, conhecidas como drusas, que se acumulam abaixo do epitélio pigmentar da retina (a camada pigmentada da retina). Com o tempo, as drusas podem crescer e se unir, criando um padrão de favo de mel. Geralmente começa no início da idade adulta, mas a idade de início varia. O grau de perda de visão também varia. A DHRD geralmente é causada por mutações no gene EFEMP1 e é herdada de maneira autossômica dominante.
Introdução
O que você precisa saber de cara
A distrofia retiniana em favo de mel de Doyne (DHRD) é uma condição que afeta os olhos e causa perda de visão. É caracterizada por pequenas manchas brancas, redondas, conhecidas como drusas, que se acumulam abaixo do epitélio pigmentar da retina (a camada pigmentada da retina). Com o tempo, as drusas podem crescer e se unir, criando um padrão de favo de mel. Geralmente começa no início da idade adulta, mas a idade de início varia. O grau de perda de visão também varia. A DHRD geralmente é causada por mutações no gene EFEMP1 e é herdada de maneira autossômica dominante.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 6 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 23 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
3 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisBinds EGFR, the EGF receptor, inducing EGFR autophosphorylation and the activation of downstream signaling pathways. May play a role in cell adhesion and migration. May function as a negative regulator of chondrocyte differentiation. In the olfactory epithelium, it may regulate glial cell migration, differentiation and the ability of glial cells to support neuronal neurite outgrowth
Secreted, extracellular space, extracellular matrix
Doyne honeycomb retinal dystrophy
An autosomal dominant, progressive, ocular disorder characterized by yellow-white deposits known as drusen that accumulate beneath the retinal pigment epithelium. With age, drusen increase in size and number, and eventually cause visual symptoms, including decreased visual acuity, metamorphopsia, photophobia, and paracentral scotoma.
Glycoprotein that plays an essential role in maintaining a well-balanced immune response by modulating complement activation. Acts as a soluble inhibitor of complement, where its binding to self markers such as glycan structures prevents complement activation and amplification on cell surfaces (PubMed:21285368, PubMed:21317894, PubMed:25402769). Accelerates the decay of the complement alternative pathway (AP) C3 convertase C3bBb, thus preventing local formation of more C3b, the central player of
Secreted
Basal laminar drusen
Drusen are extracellular deposits that accumulate below the retinal pigment epithelium on Bruch membrane. Basal laminar drusen refers to an early adult-onset drusen phenotype that shows a pattern of uniform small, slightly raised yellow subretinal nodules randomly scattered in the macula. In later stages, these drusen often become more numerous, with clustered groups of drusen scattered throughout the retina. In time these small basal laminar drusen may expand and ultimately lead to a serous pigment epithelial detachment of the macula that may result in vision loss.
Trypsin-like serine protease that plays an essential role in regulating the immune response by controlling all complement pathways. Inhibits these pathways by cleaving three peptide bonds in the alpha-chain of C3b and two bonds in the alpha-chain of C4b thereby inactivating these proteins (PubMed:17320177, PubMed:7360115). Essential cofactors for these reactions include factor H and C4BP in the fluid phase and membrane cofactor protein/CD46 and CR1 on cell surfaces (PubMed:12055245, PubMed:21418
Secreted, extracellular spaceSecreted
Hemolytic uremic syndrome, atypical, 3
An atypical form of hemolytic uremic syndrome. It is a complex genetic disease characterized by microangiopathic hemolytic anemia, thrombocytopenia, renal failure and absence of episodes of enterocolitis and diarrhea. In contrast to typical hemolytic uremic syndrome, atypical forms have a poorer prognosis, with higher death rates and frequent progression to end-stage renal disease.
Variantes genéticas (ClinVar)
568 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Drusas dominante
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
A review of the role of EFEMP1 in ophthalmic disease.
EGF-containing fibulin extracellular matrix protein 1 (EFEMP1), or fibulin-3, is an extracellular matrix glycoprotein encoded by the EFEMP1 gene. The role of EFEMP1 in the human eye is incompletely understood, but there are well-reported associations between mutations in the gene and a variety of ophthalmic diseases, such as myopia, juvenile open-angle glaucoma (JOAG), primary open-angle glaucoma (POAG) and familial drusen formation in Malattia Leventinese (ML)/Doyne honeycomb retinal dystrophy (DHRD). Variants which interact with EFEMP1 have also been identified in genome-wide association studies (GWAS) for age-related macular degeneration (AMD). Many of these conditions form a large component of ophthalmology case-load and have incompletely characterized pathogenesis. In this review, we will describe the role of EFEMP1 in ophthalmic disease. We discuss the role of EFEMP1 in Mendelian eye disease, its polygenic contributions to common ophthalmic conditions, and the potential to target EFEMP1 for therapeutic purposes.
Ranibizumab for the treatment of choroidal neovascularization due to cause other than age related macular degeneration.
To evaluate the safety and clinical efficacy of ranibizumab (Lucentis) in the treatment of choroidal neovascularization (CNV) caused by diseases other than age-related macular degeneration (AMD). 21 patients with mean age 61  17.2 years (min 16, max 85) with CNV due to causes other than AMD, in particular pathological myopia (n=11), angioid streaks (n=3), central serous chorioretinopathy (n=2), North Carolina macular dystrophy (n=1), dominant familial drusen (n=1) and idiopathic CNV (n=3). The patients were treated at the Ophthalmology Department of the University Hospital in Hradec Kralove with three monthly initial intravitreal injections of ranibizumab 0.5 mg with subsequent treatment regimen pro re nata (PRN). The best corrected visual acuity (BCVA) was evaluated on the ETDRS optotypes (Early Treatment Diabetic Retinopathy Study), central retinal thickness (CRT) was measured by optical coherent tomography (OCT) (Zeiss, Cirrus). These parameters were evaluated before start of the study and then at 1 (BCVA only), 4, 8, and 12 months during treatment. We also evaluated the possible occurrence of ocular and systemic side effects. Statistically significant improvement in the mean of BCVA score of 11.4 letters (p.
Late-onset night blindness with peripheral flecks accompanied by progressive trickle-like macular degeneration.
To report the clinical and genetic characteristics of 6 cases with late-onset night blindness with peripheral flecks accompanied by progressive trickle-like macular degeneration. Clinical and genetic data were collected from 6 independent patients who complained of night blindness in their fifth to eighth decade of life. The ophthalmological examinations included ophthalmoscopy, fundus autofluorescence (FAF), and full-field electroretinography (ERG). Whole exome sequencing with target gene analysis was performed to determine the causative genes and variants. All of the patients first complained of night blindness at the ages of 40-71 years. Funduscopic examinations demonstrated white or atrophic flecks scattered in the posterior pole and peripheral retina bilaterally. FAF showed patchy hypo-autofluorescence spots in the posterior pole similar to that of the trickling type of age-related macular degeneration (AMD). The region of abnormal FAF rapidly expanded with age, and one eye developed a choroidal neovascularization. The full-field scotopic ERGs with 20 min of dark adaptation were severely reduced or extinguished in all cases. There was partial recovery of the ERGs after 180 min of dark adaptation. The cone ERGs were reduced in all cases. Whole exome sequencing revealed no pathogenic variants of 301 retinal disease-associated genes. The six cases had some common features with the flecked retina syndrome, familial drusen, and late-onset retinal degeneration although none had pathogenic variants causative for these disorders. These cases may represent a subset of severe trickling AMD or a new clinical entity of acquired pan-retinal visual cycle deficiency of unknown etiology.
Publicações recentes
A review of the role of EFEMP1 in ophthalmic disease.
Ranibizumab for the treatment of choroidal neovascularization due to cause other than age related macular degeneration.
Late-onset night blindness with peripheral flecks accompanied by progressive trickle-like macular degeneration.
[Doyne retinal dystrophy--case report].
Linkage of autosomal dominant radial drusen (malattia leventinese) to chromosome 2p16-21.
📚 EuropePMC2 artigos no totalmostrando 3
A review of the role of EFEMP1 in ophthalmic disease.
Ophthalmic geneticsRanibizumab for the treatment of choroidal neovascularization due to cause other than age related macular degeneration.
Ceska a slovenska oftalmologie : casopis Ceske oftalmologicke spolecnosti a Slovenske oftalmologicke spolecnostiLate-onset night blindness with peripheral flecks accompanied by progressive trickle-like macular degeneration.
Documenta ophthalmologica. Advances in ophthalmologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Drusas dominante.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Drusas dominante
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A review of the role of EFEMP1 in ophthalmic disease.
- Ranibizumab for the treatment of choroidal neovascularization due to cause other than age related macular degeneration.Ceska a slovenska oftalmologie : casopis Ceske oftalmologicke spolecnosti a Slovenske oftalmologicke spolecnosti· 2019· PMID 31779462mais citado
- Late-onset night blindness with peripheral flecks accompanied by progressive trickle-like macular degeneration.
- [Doyne retinal dystrophy--case report].
- Linkage of autosomal dominant radial drusen (malattia leventinese) to chromosome 2p16-21.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:75376(Orphanet)
- OMIM OMIM:126600(OMIM)
- MONDO:0007471(MONDO)
- GARD:1912(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q28065548(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Drusas dominante
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata