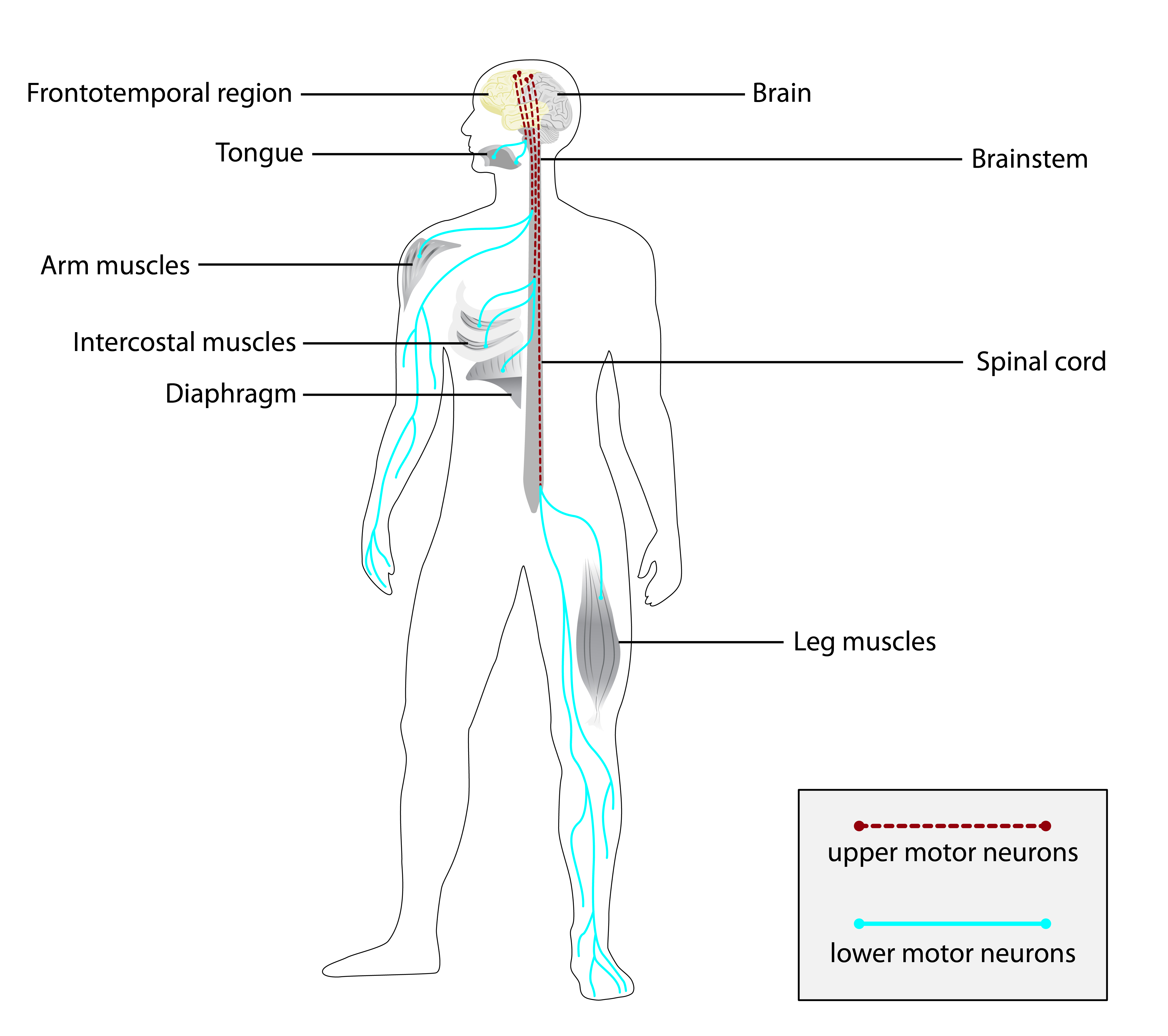
A Esclerose Lateral Primária (ELP) é uma doença neurológica que afeta os neurônios motores (as células nervosas responsáveis pelos movimentos do corpo). Ela não tem causa conhecida e não é hereditária. A doença é marcada por um problema lento e progressivo nos neurônios motores superiores, causando rigidez excessiva nos músculos (espasticidade), fraqueza leve ao fazer movimentos voluntários, reflexos exagerados (hiperreflexia) e dificuldade para falar.
Introdução
O que você precisa saber de cara
A Esclerose Lateral Primária (ELP) é uma doença neurológica que afeta os neurônios motores (as células nervosas responsáveis pelos movimentos do corpo). Ela não tem causa conhecida e não é hereditária. A doença é marcada por um problema lento e progressivo nos neurônios motores superiores, causando rigidez excessiva nos músculos (espasticidade), fraqueza leve ao fazer movimentos voluntários, reflexos exagerados (hiperreflexia) e dificuldade para falar.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 19 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 45 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
3 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, Not applicable.
Catalytic component of the m-AAA protease, a protease that plays a key role in proteostasis of inner mitochondrial membrane proteins, and which is essential for axonal and neuron development (PubMed:11549317, PubMed:28396416, PubMed:31097542, PubMed:9635427). SPG7 possesses both ATPase and protease activities: the ATPase activity is required to unfold substrates, threading them into the internal proteolytic cavity for hydrolysis into small peptide fragments (By similarity). The m-AAA protease ex
Mitochondrion inner membrane
Spastic paraplegia 7, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG7 is a complex form. Additional clinical features are cerebellar syndrome, supranuclear palsy, and cognitive impairment, particularly disturbance of attention and executive functions.
May act as a GTPase regulator. Controls survival and growth of spinal motoneurons (By similarity)
Amyotrophic lateral sclerosis 2
A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases.
Component of the ERLIN1/ERLIN2 complex which mediates the endoplasmic reticulum-associated degradation (ERAD) of inositol 1,4,5-trisphosphate receptors (IP3Rs) such as ITPR1 (PubMed:17502376, PubMed:19240031). Promotes sterol-accelerated ERAD of HMGCR probably implicating an AMFR/gp78-containing ubiquitin ligase complex (PubMed:21343306). Involved in regulation of cellular cholesterol homeostasis by regulation the SREBP signaling pathway. May promote ER retention of the SCAP-SREBF complex (PubMe
Endoplasmic reticulum membrane
Spastic paraplegia 18B, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG18B is a severe form with onset in early childhood. Most affected individuals have severe psychomotor retardation. Some may develop significant joint contractures.
Variantes genéticas (ClinVar)
733 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 32 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
9 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Esclerose lateral primária
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
20 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
The Amyotrophic Lateral Sclerosis House Call Program: A Single-Center Experience in the United States.
Accessing multidisciplinary care poses challenges for people living with amyotrophic lateral sclerosis (ALS) due to mobility issues. As ALS care rarely requires hospital-based technology, most care is available through home visits. The Daniella Lipper ALS House Call Program (HCP) at Massachusetts General Hospital (MGH), launched in 2017 in collaboration with Compassionate Care ALS, has pioneered home-based ALS care in Eastern Massachusetts. A retrospective chart review of ALS and primary lateral sclerosis (PLS) patients enrolled in the HCP at MGH was conducted. Data on demographics, visit details, and procedures performed during home visits were collected from electronic health records for patients seen from January 2024 to December 2024. In 2024, the ALS HCP conducted 959 visits for 142 patients (average age: 68 years, range: 36-93; 47.9% female). Of these patients, 137 (96.5%) were diagnosed with ALS and 5 (3.5%) with PLS. Notably, 61 patients (43%) received care exclusively at home. Key interventions included 44 gastrostomy tube exchanges and 59 respiratory assessments, both of which significantly reduced hospital visits. The average distance traveled by the care team was 30.32 miles per visit. The Daniella Lipper ALS HCP at MGH brings ALS expertise into the patient's home, minimizing travel burdens and ensuring continuity of care. The program illustrates the feasibility and impact of home-based ALS care, suggesting potential for broader implementation across the nation. Development will focus on expanding services, such as tracheostomy changes in the homes, and on creating sustainable models for similar initiatives.
TBK1-Associated Primary Lateral Sclerosis Followed by Right Temporal Variant Frontotemporal Dementia.
We report a 58-year-old woman with a novel splice-site variant in the TANK-binding kinase 1 (TBK1:c.993-2A>C p.Ala332TyrfsTer39) who sequentially developed primary lateral sclerosis (PLS) followed by right temporal variant frontotemporal dementia (rtvFTD). Neuroimaging demonstrated right anterior temporal atrophy before cognitive symptoms, and prosopagnosia represented the earliest manifestation of rtvFTD. Molecular analysis revealed reduced levels of correctly spliced TBK1 transcripts, consistent with haploinsufficiency. Given the shared involvement of TDP-43 pathology in both PLS and rtvFTD, this case indicates TBK1 dysfunction as a fundamental genetic factor underlying the coexistence of these phenotypes, underscoring the clinical value of early neuroimaging and genetic evaluation.
Demographic, clinical and genetic characteristics of patients with amyotrophic lateral sclerosis from two specialised centres in Austria.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterised by progressive muscle weakness and ultimately death from respiratory failure. Heterogeneity in disease trajectories and outcomes among patients with ALS (pwALS) is influenced by healthcare access, rehabilitation, and palliative care, but real-world data on demographic and clinical characteristics remain scarce in many countries, including Austria. To characterise the demographic, clinical, and genetic landscape of pwALS in Austria. In this retrospective cohort study, we included pwALS diagnosed according to the Gold Coast criteria and treated at two large tertiary referral centres. Demographic, clinical, and genetic data were extracted from the local ALS registries, and survival was determined via linkage with Statistik Austria, censored in December 2023. A total of 341 patients with motor neuron disease were included (44.9% female), of whom 5% were diagnosed with primary lateral sclerosis and 2.9% with progressive muscular atrophy. Among pwALS (n = 314), spinal onset was most common (67.2%), followed by bulbar onset (29.6%) and respiratory onset (2.5%). Median survival from symptom onset was 36.0 months (IQR 20.0-74.0), with age at onset (HR 1.04, 95% CI 1.02-1.05; p < 0.0001), diagnostic delay (HR 0.97, 95% CI 0.96-0.98; p < 0.0001), and PEG tube placement (HR 0.72, 95% CI 0.50-1.00; p = 0.0478) as the only independent predictors of survival. (Likely) pathogenic variants were identified in 5.5% of patients, including two in SOD1 and one each in C9orf72, OPTN, TARDBP, and FUS. This study provides the first comprehensive description of the demographic, clinical, and genetic characteristics of pwALS in Austria, offering valuable real-world insight into disease presentation and genetic diversity.
The Epidemiology of Primary Lateral Sclerosis: Results from a Population-Based Cohort.
In this population-based study, we described the epidemiology of primary lateral sclerosis (PLS) in northern Italy and compared the clinical characteristics of patients with PLS to those with predominant upper motor neuron (PUMN) involvement and classic amyotrophic lateral sclerosis (ALS). Patients from the PARALS registry diagnosed with probable or definite PLS between 2007 and 2021 were included. Crude annual incidence rates were calculated, along with age- and sex-specific rates. A survival analysis was performed to identify prognostic factors at diagnosis. Covariates included sex, age at onset, site of onset, diagnostic delay, forced vital capacity (FVC), change in ALS Functional Rating Scale (ΔFRS), and change in body mass index (ΔBMI). A total of 57 PLS patients (2.7%) were included, with a crude incidence rate of 0.084 per 100,000 person-years. Compared to PUMN and classic ALS, PLS patients were younger (median onset age 63.5 years, interquartile range [IQR] 54.9-70.4) and predominantly female (male-to-female ratio 0.58). Bulbar onset occurred in 11 cases (19.3%), all of whom later developed spinal symptoms. At censoring, 38 patients (66.7%) were still alive (median survival 8.3 years, IQR 5.7-12.3), corresponding to a point prevalence of 0.89 per 100,000. Survival was significantly associated with age at onset (hazard ratio [HR] 1.17, 95% confidence interval [CI]: 1.05-1.33, p = 0.001), male sex (HR 4.41, 95% CI: 1.24-15.6, p = 0.02), and FVC at diagnosis (HR 0.95, 95% CI: 0.93-0.98, p = 0.006). PLS was confirmed to be rarer than other ALS phenotypes. Patients had a higher age at onset than previously reported and a female predominance. Sex, age at onset, and respiratory function were key prognostic factors. ANN NEUROL 2026;99:606-613.
Primary Lateral Sclerosis Natural History Study: Primary Lateral Sclerosis Functional Rating Scale and Other Outcomes Assessment.
The primary lateral sclerosis (PLS) consensus diagnostic criteria and functional rating scale (PLSFRS) were recently established to facilitate and optimize future PLS clinical trials. We examined the trajectory of the PLSFRS and other functional outcome measures and biomarkers in the PLS Natural History Study (PLS NHS) to understand their performance in this prospective cohort. The PLS NHS is a prospective, longitudinal, multicenter study of people living with PLS in different diagnostic categories: early (disease duration <2 years); probable (2-4 years); and definite PLS (4-15 years). PLSFRS scores and other functional outcome measures were collected at baseline, 3-, 6-, 9-, and 12-month follow-up visits. Baseline characteristics were compared between the groups. The slopes of the PLSFRS and other functional outcome measures over 12 months were examined in the overall cohort and subgroups using linear mixed-effect models. The associations between baseline characteristics and the rate of PLSFRS decline were analyzed with linear regression models. A total of 76 participants were included: early (n = 6); probable (n = 26); and definite (n = 44) PLS. Baseline PLSFRS total scores were highest in the early PLS group, followed by the probable and definite PLS groups. In the overall cohort, the PLSFRS total score declined by 0.33 points/month (95% confidence interval [0.27-0.39], adjusted p < 0.05). The rate of decline was steepest in the early PLS group, followed by the probable and definite PLS groups. Baseline neurofilament light chain level was associated with the rate of PLSFRS decline over 1 year (p = 0.001). In PLS, the rate of functional decline, as measured by the PLSFRS total score, is faster during the early phase of the disease. Neurofilament light might serve as a prognostic biomarker in PLS. ANN NEUROL 2026;99:418-428.
Publicações recentes
Primary lateral sclerosis in Brazil: phenotypic heterogeneity, non-motor features, and prognostic markers in a 17-year multicentre cohort.
Disappearing corticospinal tract on routine MRI: dynamic signal evolution in primary lateral sclerosis.
Neuroimaging confirms selective cerebral involvement in primary lateral sclerosis and predilection to brain regions with high metabolic activity.
Expanding the Motor Band Sign in Motor Neuron Disease Using 7T MRI: Visualization of Cortical Layer-Dependent Iron Deposition in the Primary Motor Cortex.
Prospective Validation of the New PLS Diagnostic Criteria From PLS Natural History Study: EMG and Neurofilament Analyses.
📚 EuropePMC269 artigos no totalmostrando 193
Expanding the Motor Band Sign in Motor Neuron Disease Using 7T MRI: Visualization of Cortical Layer-Dependent Iron Deposition in the Primary Motor Cortex.
Muscle & nerveProspective Validation of the New PLS Diagnostic Criteria From PLS Natural History Study: EMG and Neurofilament Analyses.
Muscle & nerveThe Amyotrophic Lateral Sclerosis House Call Program: A Single-Center Experience in the United States.
Neurology research internationalTrajectory of Mobility Function Decline for People With Motor Neuron Disease.
Archives of physical medicine and rehabilitationTBK1-Associated Primary Lateral Sclerosis Followed by Right Temporal Variant Frontotemporal Dementia.
Annals of clinical and translational neurologyInvolvement of Keap1/Nrf2 and the antioxidant defence in cytoprotective effects induced by cannabis polyphenols in SH-SY5Y neuronal cells.
Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapieRiluzole use and reasons for non-use in people with amyotrophic lateral sclerosis in Aotearoa New Zealand.
The New Zealand medical journalEstablishing Diagnostic and Differential Diagnostic Criteria for Amyotrophic Lateral Sclerosis.
Journal of clinical medicineDemographic, clinical and genetic characteristics of patients with amyotrophic lateral sclerosis from two specialised centres in Austria.
Journal of neurologyCo-aggregation of annexin A11 and TDP-43 in FTLD/MND with primary lateral sclerosis phenotype.
Acta neuropathologica communicationsVariably Protease-Sensitive Prionopathy: Two New Cases With Motor Neuron-Dementia Syndrome.
Annals of clinical and translational neurologyMotor band sign in 18F-FDG PET/CT studies: a biomarker of degenerative upper motor neuron disease? A study of three cases and literature review.
NeurologiaCase Series Assessing the Use of Levetiracetam for Gait Improvement in Primary Lateral Sclerosis.
The Canadian journal of neurological sciences. Le journal canadien des sciences neurologiquesStartle Reflex in Primary Lateral Sclerosis (PLS): A Comparison With Amyotrophic Lateral Sclerosis (ALS).
Muscle & nerveThe Epidemiology of Primary Lateral Sclerosis: Results from a Population-Based Cohort.
Annals of neurologyThe genetic architecture of primary lateral sclerosis in a cohort of Italian patients.
Journal of neurologyExpanding the spectrum of annexin A11 proteinopathy in frontotemporal lobar degeneration and motor neuron disease.
Acta neuropathologicaSpanish adaptation of the Primary Lateral Sclerosis Functional Rating Scale (PLSFRS).
Amyotrophic lateral sclerosis & frontotemporal degenerationMotor Cortex Coverage Predicts Signal Strength of a Stentrode Endovascular Brain-Computer Interface.
medRxiv : the preprint server for health sciencesDevelopment of a Human iPSC-Derived "Corticospinal Tract-on-a-Chip" for Neurodegenerative Disease Research.
bioRxiv : the preprint server for biologyPrimary Lateral Sclerosis Natural History Study: Primary Lateral Sclerosis Functional Rating Scale and Other Outcomes Assessment.
Annals of neurologyAtypical features including acquired oculomotor apraxia in C9orf72-associated familial primary lateral sclerosis.
Journal of neuromuscular diseasesSerum NfL, but not GFAP, differentiates primary lateral sclerosis from adrenomyeloneuropathy and hereditary spastic paraplegia type 4.
Amyotrophic lateral sclerosis & frontotemporal degenerationGeneration and characterization of two pluripotent stem cell lines from Primary Lateral Sclerosis (PLS) patients.
Stem cell researchAmyotrophic Lateral Sclerosis Masquerading as Multiple System Atrophy with Parkinsonism and Anxiety as Initial Manifestations.
Degenerative neurological and neuromuscular diseaseWhole genome sequencing analysis in primary lateral sclerosis (PLS) patients reveals mutations in neurological diseases-causing genes.
Journal of neurologyA Japanese familial spastic paraplegia associated with a missense UBQLN2 variant.
Journal of human geneticsUnilateral Motor Band Sign in Hemiplegic Primary Lateral Sclerosis.
Neurology IndiaNeuroaxonal Degeneration as a Converging Mechanism in Motor Neuron Diseases (MNDs): Molecular Insights into RNA Dysregulation and Emerging Therapeutic Targets.
International journal of molecular sciencesToward therapeutic trials in primary lateral sclerosis.
Amyotrophic lateral sclerosis & frontotemporal degenerationCortical Excitability as a Prognostic and Phenotypic Stratification Biomarker in Amyotrophic Lateral Sclerosis.
Annals of neurologyWhat Is in the Literature.
Journal of clinical neuromuscular diseaseBurden of pathogenetic and likely pathogenetic variants in SPG7, SPG11 and AP4 genes in Amyotrophic Lateral Sclerosis. A case-control study.
Journal of neurologyDifferentiating upper- and lower motor neuron diseases using automated acoustic analysis.
Amyotrophic lateral sclerosis & frontotemporal degenerationInconsistent primary motor cortex glucose hypometabolism in primary lateral sclerosis.
Journal of neurologyPrimary Lateral Sclerosis: Implications for Diagnostic Criteria From a Natural History Study in the Netherlands.
NeurologyPrimary Lateral Sclerosis (Progressive Spinobulbar Spasticity): Serial Analysis Over Many Years Increases Diagnostic Certainty.
NeurologyProgression and life expectancy in primary lateral sclerosis.
Journal of neurology, neurosurgery, and psychiatryLatest progress and challenges in drug development for degenerative motor neuron diseases.
Neural regeneration researchNew developments in imaging in ALS.
Journal of neurology[Genetics of Motor Neuron Diseases and Hereditary Spastic Paraplegia].
Brain and nerve = Shinkei kenkyu no shinpoAdvances and research priorities in the respiratory management of ALS: Historical perspectives and new technologies.
Revue neurologiquePathological laugher and crying in motor neuron diseases: a matter of bulbar and neurobehavioral involvement with sex imbalance.
Journal of neurologyMotor phenotypes of amyotrophic lateral sclerosis - a three-determinant anatomical classification based on the region of onset, propagation of motor symptoms, and the degree of upper and lower motor neuron dysfunction.
Neurological research and practiceTDP-43 seeding activity in the olfactory mucosa of patients with amyotrophic lateral sclerosis.
Molecular neurodegenerationSensory Dysfunction in ALS and Other Motor Neuron Diseases: Clinical Relevance, Histopathology, Neurophysiology, and Insights from Neuroimaging.
BiomedicinesPseudobulbar affect: clinical associations, social impact and quality of life implications - Lessons from PLS.
Journal of neurologyHIV associated motor neuron disease (MND): A case series with systematic review of literature.
Journal of neurovirologyCervical Collar Satisfaction and Functional Impact in Amyotrophic Lateral Sclerosis: A Survey Study.
American journal of physical medicine & rehabilitationThe Role of Gastrostomy and Noninvasive Ventilation in Primary Lateral Sclerosis.
Muscle & nerveHypothalamic atrophy in primary lateral sclerosis, assessed by convolutional neural network-based automatic segmentation.
Scientific reportsAssociation between cardiometabolic diseases and the risk and progression of motor neuron diseases in Sweden: a population-based case-control study.
The Lancet regional health. EuropeHigh-density multielectrode arrays bring cellular resolution to neuronal activity and network analyses of corticospinal motor neurons.
Scientific reportsDiagnosing primary lateral sclerosis: a clinico-pathological study.
Journal of neurologyMotor Neuron Diseases and Central Nervous System Tractopathies: Clinical-Radiologic Correlation and Diagnostic Approach.
Radiographics : a review publication of the Radiological Society of North America, IncDevelopment and validation a novel FEZF2 based fluorescent reporter for corticospinal motor neurons.
Metabolic brain diseaseUtilizing quantitative susceptibility mapping to differentiate primary lateral sclerosis from progressive supranuclear palsy: A case report.
Neuropathology : official journal of the Japanese Society of NeuropathologyAmyotrophic lateral sclerosis established as a multistep process across phenotypes.
European journal of neurologyPhenotype and Genotype of Children with ALS2 gene-Related Disorder.
NeuropediatricsSafety and efficacy of memantine and trazodone versus placebo for motor neuron disease (MND SMART): stage two interim analysis from the first cycle of a phase 3, multiarm, multistage, randomised, adaptive platform trial.
The Lancet. NeurologyA microRNA diagnostic biomarker for amyotrophic lateral sclerosis.
Brain communicationsLongitudinal analysis of glymphatic function in amyotrophic lateral sclerosis and primary lateral sclerosis.
Brain : a journal of neurologyComparative analysis of neurofilaments and biomarkers of muscular damage in amyotrophic lateral sclerosis.
Brain communicationsAn autopsy case of progressive supranuclear palsy with severe corticospinal tract degeneration.
Neuropathology : official journal of the Japanese Society of NeuropathologyReliability and consistency of the Japanese version of the Primary Lateral Sclerosis Functional Rating Scale.
BMC neurologyPrimary lateral sclerosis associated with PSEN1 Pro284Leu variant in a Colombian family: Clinical and neuropathological features.
Alzheimer's & dementia : the journal of the Alzheimer's AssociationUpper motor neuron-predominant motor neuron disease presenting as atypical parkinsonism: A clinicopathological study.
Brain pathology (Zurich, Switzerland)Decoding genetic and pathophysiological mechanisms in amyotrophic lateral sclerosis and primary lateral sclerosis: A comparative study of differentially expressed genes and implicated pathways in motor neuron disorders.
Advances in protein chemistry and structural biologyRare variant analyses validate known ALS genes in a multi-ethnic population and identifies ANTXR2 as a candidate in PLS.
BMC genomicsAnalysis and occurrence of biallelic pathogenic repeat expansions in RFC1 in a German cohort of patients with a main clinical phenotype of motor neuron disease.
Journal of neurologyAutonomic impairment in primary lateral sclerosis.
Clinical autonomic research : official journal of the Clinical Autonomic Research SocietySerum neurofilament light chain in distinct phenotypes of amyotrophic lateral sclerosis: A longitudinal, multicenter study.
European journal of neurologyThe periprocedural respiratory safety of propofol sedation in patients with a motor neuron disease undergoing percutaneous endoscopic gastrostomy insertion.
Journal of the neurological sciencesCould PLS represent a UMN-predominant ALS syndrome?
Revue neurologiqueAcceptance and Commitment Therapy plus usual care for improving quality of life in people with motor neuron disease (COMMEND): a multicentre, parallel, randomised controlled trial in the UK.
Lancet (London, England)Upper motor neuron signs in primary lateral sclerosis and hereditary spastic paraplegia.
Muscle & nerveClinical and neuroimaging characteristics of primary lateral sclerosis with overlapping features of progressive supranuclear palsy.
European journal of neurologyRepeat expansions in AR, ATXN1, ATXN2 and HTT in Norwegian patients diagnosed with amyotrophic lateral sclerosis.
Brain communicationsHarnessing Big Data in Amyotrophic Lateral Sclerosis: Machine Learning Applications for Clinical Practice and Pharmaceutical Trials.
Journal of integrative neuroscienceWhole-body fasciculation detection in amyotrophic lateral sclerosis using motor unit MRI.
Clinical neurophysiology : official journal of the International Federation of Clinical NeurophysiologySupra- and infra-tentorial degeneration patterns in primary lateral sclerosis: a multimodal longitudinal neuroradiology study.
Journal of neurologyPrimary lateral sclerosis: application and validation of the 2020 consensus diagnostic criteria in an expert opinion-based PLS cohort.
Journal of neurology, neurosurgery, and psychiatryPhenotypic Spectrum of Progressive Supranuclear Palsy: Clinical Study and Apolipoprotein E Effect.
Journal of movement disordersPrimary Lateral Sclerosis: An Overview.
Journal of clinical medicinePrimary lateral sclerosis: more than just an upper motor neuron disease.
Neural regeneration researchCurrent challenges in primary lateral sclerosis diagnosis.
Expert review of neurotherapeuticsUpper Limb Movement Execution Classification using Electroencephalography for Brain Computer Interface.
Annual International Conference of the IEEE Engineering in Medicine and Biology Society. IEEE Engineering in Medicine and Biology Society. Annual International ConferenceA case of Mills' syndrome: initially characterized by one cerebral hemisphere atrophy and decreased brain metabolism then evolving into amyotrophic lateral sclerosis.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyKetamine-Assisted Psychodynamic Psychotherapy.
Psychodynamic psychiatryAn autopsy case of diffuse atypical argyrophilic grain disease (AGD) with presenile onset and three-year course of motor and cognitive impairment.
Neuropathology : official journal of the Japanese Society of NeuropathologyLanguage deficits in primary lateral sclerosis: cortical atrophy, white matter degeneration and functional disconnection between cerebral regions.
Journal of neurologyFamilial motor neuron disease: co-occurrence of PLS and ALS (-FTD).
Amyotrophic lateral sclerosis & frontotemporal degenerationPrimary lateral sclerosis plus parkinsonism: a case report.
BMC neurologyPrimary lateral sclerosis.
Handbook of clinical neurologyThe hereditary spastic paraplegias.
Handbook of clinical neurologyCreatine Kinase MB Isoenzyme Is a Complementary Biomarker in Amyotrophic Lateral Sclerosis.
International journal of molecular sciencesRadiological correlates of pseudobulbar affect: Corticobulbar and cerebellar components in primary lateral sclerosis.
Journal of the neurological sciencesSerum Neurofilaments in Motor Neuron Disease and Their Utility in Differentiating ALS, PMA and PLS.
Life (Basel, Switzerland)Role of the immune system in amyotrophic lateral sclerosis. Analysis of the natural killer cells and other circulating lymphocytes in a cohort of ALS patients.
BMC neurologyLimited value of serum neurofilament light chain in diagnosing amyotrophic lateral sclerosis.
Brain communicationsAdvantages of routine next-generation sequencing over standard genetic testing in the amyotrophic lateral sclerosis clinic.
European journal of neurologyCortico-muscular coherence in primary lateral sclerosis reveals abnormal cortical engagement during motor function beyond primary motor areas.
Cerebral cortex (New York, N.Y. : 1991)Genetic characterization of primary lateral sclerosis.
Journal of neurologyRegional spreading pattern is associated with clinical phenotype in amyotrophic lateral sclerosis.
Brain : a journal of neurologyMotor Band Sign in Primary Lateral Sclerosis: A Case Report Proposing the Need for an Imaging Biomarker.
CureusClinical and molecular spectrum of a large Egyptian cohort with ALS2-related disorders of infantile-onset of clinical continuum IAHSP/JPLS.
Clinical geneticsPrimary Lateral Sclerosis: Can Rocuronium Be an Option?
CureusPhenotypic correlates of serum neurofilament light chain levels in amyotrophic lateral sclerosis.
Frontiers in aging neuroscienceUtilizing machine learning and lipidomics to distinguish primary lateral sclerosis from amyotrophic lateral sclerosis.
Muscle & nerveNot a benign motor neuron disease: longitudinal imaging captures relentless motor connectome disintegration in primary lateral sclerosis.
European journal of neurologyProgress and challenges in directing the differentiation of human iPSCs into spinal motor neurons.
Frontiers in cell and developmental biologyM1/precuneus ratio as a surrogate marker of upper motor neuron sign in ALS.
Journal of the neurological sciencesAssessment of Safety of a Fully Implanted Endovascular Brain-Computer Interface for Severe Paralysis in 4 Patients: The Stentrode With Thought-Controlled Digital Switch (SWITCH) Study.
JAMA neurologyPrimary lateral sclerosis natural history study - planning, designing, and early enrollment.
Amyotrophic lateral sclerosis & frontotemporal degenerationGenotype-phenotype characterisation of long survivors with motor neuron disease in Scotland.
Journal of neurologyGlobular glial tauopathy type II.
Practical neurologyCurrent state of research on acupuncture for the treatment of amyotrophic lateral sclerosis: A scoping review.
Frontiers in neurologyA randomised controlled trial of acceptance and commitment therapy plus usual care compared to usual care alone for improving psychological health in people with motor neuron disease (COMMEND): study protocol.
BMC neurologyProgressive motor neuron syndromes with single CNS lesions and CSF oligoclonal bands: never forget solitary sclerosis!
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyCase report: A variant of the FIG4 gene with rapidly progressive amyotrophic lateral sclerosis.
Frontiers in neurologyMills' syndrome is a unique entity of upper motor neuron disease with N-shaped progression: Three case reports.
World journal of clinical casesClinical Spectrum of Tauopathies.
Frontiers in neurologyParkinsonian Syndromes in Motor Neuron Disease: A Clinical Study.
Frontiers in aging neuroscienceNonalcoholic Fatty Liver Disease in Patients with Inherited and Sporadic Motor Neuron Degeneration.
GenesSegmental alterations of the corpus callosum in motor neuron disease: A DTI and texture analysis in 575 patients.
NeuroImage. ClinicalIron-sensitive MR imaging of the primary motor cortex to differentiate hereditary spastic paraplegia from other motor neuron diseases.
European radiologyCerebellar pathology in motor neuron disease: neuroplasticity and neurodegeneration.
Neural regeneration researchThe Global Burden of Motor Neuron Disease: An Analysis of the 2019 Global Burden of Disease Study.
Frontiers in neurologyArthroscopic Lateral Collateral Ligament Repair for the Acute Elbow Dislocation in Primary Lateral Sclerosis Patient: A Case Report.
Orthopedic reviewsMotor Band Sign in Motor Neuron Disease: A Marker for Upper Motor Neuron Involvement.
The Canadian journal of neurological sciences. Le journal canadien des sciences neurologiquesAcoustic Change Over Time in Spastic and/or Flaccid Dysarthria in Motor Neuron Diseases.
Journal of speech, language, and hearing research : JSLHRParental and child adjustment to amyotrophic lateral sclerosis: transformations, struggles and needs.
BMC psychologyPrimary lateral sclerosis: diagnostic contribution of brain [18F]FDG PET/CT.
Revista espanola de medicina nuclear e imagen molecularFrontotemporal Dementia as a Possible Manifestation of Primary Lateral Sclerosis: A Case Report and Literature Review.
Case reports in psychiatryDifferential Diagnosis of Rare Subtypes of Progressive Supranuclear Palsy and PSP-Like Syndromes-Infrequent Manifestations of the Most Common Form of Atypical Parkinsonism.
Frontiers in aging neuroscienceDoes limited EMG denervation in early primary lateral sclerosis predict amyotrophic lateral sclerosis?
Amyotrophic lateral sclerosis & frontotemporal degenerationMotor neuron disease in three asymptomatic pVal50Met TTR gene carriers.
Amyotrophic lateral sclerosis & frontotemporal degeneration[Oral hygiene in patients with motor neuron disease: a cross-sectional survey].
Nederlands tijdschrift voor tandheelkundePrimary Lateral Sclerosis Presenting With Focal Onset Spreading Through Contiguous Neuroanatomic Regions.
NeurologyHome-monitoring of vital capacity in people with a motor neuron disease.
Journal of neurologyAsymmetrical Primary Lateral Sclerosis Presenting as Corticobasal Syndrome.
Journal of neuropathology and experimental neurologyALS2-Related Motor Neuron Diseases: From Symptoms to Molecules.
BiologyCerebellar degeneration in primary lateral sclerosis: an under-recognized facet of PLS.
Amyotrophic lateral sclerosis & frontotemporal degenerationA Case Report on Robot-Aided Gait Training in Primary Lateral Sclerosis Rehabilitation: Rationale, Feasibility and Potential Effectiveness of a Novel Rehabilitation Approach.
Innovations in clinical neuroscienceAI-based protein structure databases have the potential to accelerate rare diseases research: AlphaFoldDB and the case of IAHSP/Alsin.
Drug discovery todayImaging data reveal divergent longitudinal trajectories in PLS, ALS and poliomyelitis survivors: Group-level and single-subject traits.
Data in briefPhenotypic categorisation of individual subjects with motor neuron disease based on radiological disease burden patterns: A machine-learning approach.
Journal of the neurological sciencesUpper motor neurons are a target for gene therapy and UCHL1 is necessary and sufficient to improve cellular integrity of diseased upper motor neurons.
Gene therapyCSF Heavy Neurofilament May Discriminate and Predict Motor Neuron Diseases with Upper Motor Neuron Involvement.
BiomedicinesHalf Wine Glass Appearance in Early Primary Lateral Sclerosis.
Neurology IndiaAmyotrophic lateral sclerosis (ALS) and the endocrine system: Are there any further ties to be explored?
Aging brainThe expanding clinical and genetic spectrum of alsin-related disorders: the first cohort of Brazilian patients.
Amyotrophic lateral sclerosis & frontotemporal degenerationElectromyographic findings in primary lateral sclerosis during disease progression.
Clinical neurophysiology : official journal of the International Federation of Clinical NeurophysiologyFamilial clustering of primary lateral sclerosis and amyotrophic lateral sclerosis: Supplementary evidence for a continuum.
European journal of neurologyEmotional Processing and Experience in Amyotrophic Lateral Sclerosis: A Systematic and Critical Review.
Brain sciencesPropagation patterns in motor neuron diseases: Individual and phenotype-associated disease-burden trajectories across the UMN-LMN spectrum of MNDs.
Neurobiology of agingBlenderized Tube Feeding and Enterostomy Tube Occlusions Among Adults with Amyotrophic Lateral Sclerosis and Primary Lateral Sclerosis.
Canadian journal of dietetic practice and research : a publication of Dietitians of Canada = Revue canadienne de la pratique et de la recherche en dietetique : une publication des Dietetistes du CanadaPresenilin-1 Mutations Are a Cause of Primary Lateral Sclerosis-Like Syndrome.
Frontiers in molecular neuroscienceFrontotemporal Pathology in Motor Neuron Disease Phenotypes: Insights From Neuroimaging.
Frontiers in neurologyBradykinesia in motoneuron diseases.
Clinical neurophysiology : official journal of the International Federation of Clinical NeurophysiologyThe Upper Motor Neuron-Improved Knowledge from ALS and Related Clinical Disorders.
Brain sciencesRole of the nigrosome 1 absence as a biomarker in amyotrophic lateral sclerosis.
Journal of neurologyGold Coast diagnostic criteria: Implications for ALS diagnosis and clinical trial enrollment.
Muscle & nerveUtility of Transcranial Magnetic Simulation in Studying Upper Motor Neuron Dysfunction in Amyotrophic Lateral Sclerosis.
Brain sciencesCombining structural and metabolic markers in a quantitative MRI study of motor neuron diseases.
Annals of clinical and translational neurologyThe diagnostic challenge of primary lateral sclerosis: the integration of clinical, genetic and radiological cues.
European journal of neurologyInitial Cerebellar Ataxia in Hereditary Adult-Onset Primary Lateral Sclerosis.
Case reports in neurologyPrimary Lateral Sclerosis: Clinical, radiological and molecular features.
Revue neurologiqueLipidomics study of plasma from patients suggest that ALS and PLS are part of a continuum of motor neuron disorders.
Scientific reportsSMN1 Duplications Are Associated With Progressive Muscular Atrophy, but Not With Multifocal Motor Neuropathy and Primary Lateral Sclerosis.
Neurology. GeneticsUpper Motor Neuron Disorders: Primary Lateral Sclerosis, Upper Motor Neuron Dominant Amyotrophic Lateral Sclerosis, and Hereditary Spastic Paraplegia.
Brain sciencesPrevalence of multimorbidity and its impact on survival in people with motor neuron disease.
European journal of neurologyDistinguishing Primary Lateral Sclerosis from Parkinsonian Syndromes with the Help of Advanced Imaging.
Journal of nuclear medicine : official publication, Society of Nuclear MedicineMutations and Protein Interaction Landscape Reveal Key Cellular Events Perturbed in Upper Motor Neurons with HSP and PLS.
Brain sciencesEstimation of the prevalence and incidence of motor neuron diseases in two Spanish regions: Catalonia and Valencia.
Scientific reportsNatural History of "Pure" Primary Lateral Sclerosis.
NeurologyImproving mitochondria and ER stability helps eliminate upper motor neuron degeneration that occurs due to mSOD1 toxicity and TDP-43 pathology.
Clinical and translational medicineClinical care and therapeutic trials in PLS.
Amyotrophic lateral sclerosis & frontotemporal degenerationMeasuring disease progression in primary lateral sclerosis.
Amyotrophic lateral sclerosis & frontotemporal degenerationNeuroimaging in primary lateral sclerosis.
Amyotrophic lateral sclerosis & frontotemporal degenerationBetter understanding the neurobiology of primary lateral sclerosis.
Amyotrophic lateral sclerosis & frontotemporal degenerationThe clinical spectrum of primary lateral sclerosis.
Amyotrophic lateral sclerosis & frontotemporal degenerationGenetics of primary lateral sclerosis.
Amyotrophic lateral sclerosis & frontotemporal degenerationNeurophysiological features of primary lateral sclerosis.
Amyotrophic lateral sclerosis & frontotemporal degenerationNeuropathology of primary lateral sclerosis.
Amyotrophic lateral sclerosis & frontotemporal degenerationDiagnostics of Amyotrophic Lateral Sclerosis: Up to Date.
Diagnostics (Basel, Switzerland)Progression of cognitive and behavioral disturbances in motor neuron diseases assessed using standard and computer-based batteries.
Amyotrophic lateral sclerosis & frontotemporal degenerationCase Report: Association of a Variant of Unknown Significance in the FIG4 Gene With Frontotemporal Dementia and Slowly Progressing Motoneuron Disease: A Case Report Depicting Common Challenges in Clinical and Genetic Diagnostics of Rare Neuropsychiatric and Neurologic Disorders.
Frontiers in neuroscienceExtra-motor cerebral changes and manifestations in primary lateral sclerosis.
Brain imaging and behaviorThe cutaneous silent period in motor neuron disease.
Clinical neurophysiology : official journal of the International Federation of Clinical NeurophysiologyA case of vertebral artery compression syndrome mimicking primary lateral sclerosis.
The International journal of neuroscienceImportance of lipids for upper motor neuron health and disease.
Seminars in cell & developmental biologyThe Importance of Personally Reviewing Imaging for Clinical Correlation: A Case of Thoracic Spinal Stenosis Masquerading as a Motor Neuron Disease.
American journal of physical medicine & rehabilitationAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Esclerose lateral primária.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Esclerose lateral primária
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- The Amyotrophic Lateral Sclerosis House Call Program: A Single-Center Experience in the United States.
- TBK1-Associated Primary Lateral Sclerosis Followed by Right Temporal Variant Frontotemporal Dementia.
- Demographic, clinical and genetic characteristics of patients with amyotrophic lateral sclerosis from two specialised centres in Austria.
- The Epidemiology of Primary Lateral Sclerosis: Results from a Population-Based Cohort.
- Primary Lateral Sclerosis Natural History Study: Primary Lateral Sclerosis Functional Rating Scale and Other Outcomes Assessment.
- Primary lateral sclerosis in Brazil: phenotypic heterogeneity, non-motor features, and prognostic markers in a 17-year multicentre cohort.
- Disappearing corticospinal tract on routine MRI: dynamic signal evolution in primary lateral sclerosis.
- Neuroimaging confirms selective cerebral involvement in primary lateral sclerosis and predilection to brain regions with high metabolic activity.
- Expanding the Motor Band Sign in Motor Neuron Disease Using 7T MRI: Visualization of Cortical Layer-Dependent Iron Deposition in the Primary Motor Cortex.
- Prospective Validation of the New PLS Diagnostic Criteria From PLS Natural History Study: EMG and Neurofilament Analyses.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:35689(Orphanet)
- MONDO:0018155(MONDO)
- GARD:10684(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q2881413(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Esclerose lateral primária
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov