A Neoplasia Endócrina Múltipla tipo 4 (MEN4) é uma forma muito rara da síndrome MEN, que é uma condição de câncer hereditária (passada de geração em geração). Ela se caracteriza por tumores nas glândulas paratireoides e na parte da frente da hipófise, podendo estar associada também a tumores nas glândulas adrenais (ou suprarrenais), nos rins e nos órgãos reprodutores.
Introdução
O que você precisa saber de cara
A Neoplasia Endócrina Múltipla tipo 4 (MEN4) é uma forma muito rara da síndrome MEN, que é uma condição de câncer hereditária (passada de geração em geração). Ela se caracteriza por tumores nas glândulas paratireoides e na parte da frente da hipófise, podendo estar associada também a tumores nas glândulas adrenais (ou suprarrenais), nos rins e nos órgãos reprodutores.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 14 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 45 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant, Not applicable.
Curadoria gene-doença
fontes oficiaisImportant regulator of cell cycle progression. Inhibits the kinase activity of CDK2 bound to cyclin A, but has little inhibitory activity on CDK2 bound to SPDYA (PubMed:28666995). Involved in G1 arrest. Potent inhibitor of cyclin E- and cyclin A-CDK2 complexes. Forms a complex with cyclin type D-CDK4 complexes and is involved in the assembly, stability, and modulation of CCND1-CDK4 complex activation. Acts either as an inhibitor or an activator of cyclin type D-CDK4 complexes depending on its ph
NucleusCytoplasmEndosome
Multiple endocrine neoplasia 4
Multiple endocrine neoplasia (MEN) syndromes are inherited cancer syndromes of the thyroid. MEN4 is a MEN-like syndrome with a phenotypic overlap of both MEN1 and MEN2.
Medicamentos aprovados (FDA)
1 medicamento encontrado nos registros da FDA americana.
Variantes genéticas (ClinVar)
259 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 863 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
16 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Neoplasia endócrina múltipla tipo 4
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
p27Kip1 and Tumors: Characterization of CDKN1B Variants Identified in MEN4 and Breast Cancer.
p27Kip1 is a key cell cycle gatekeeper governing the timing of Cyclin-dependent kinase (CDK) activation/inactivation and, consequently, cell proliferation. Structurally, the protein is largely unfolded, a feature that strongly increases its plasticity and interactors and enhances the number of regulated cellular processes. p27Kip1, like other intrinsically unstructured proteins, is post-translationally modified on several residues. These modifications affect its cellular localization and address p27Kip1 for specific interactions/functions. Several germline or somatic CDKN1B (the p27Kip1 encoding gene) mutations have been demonstrated to be associated with multiple endocrine neoplasia type 4 (MEN4), hairy cell leukemia, small-intestine neuroendocrine tumors, and breast and prostate cancers. Here, we analyzed the effect of four CDKN1B missense and nonsense mutations found in patients affected by MEN4 or cancers, namely, c.349C>T, p.P117S; c.397C>A, p.P133T; c.487C>T, p.Q163*; and c.511G>T, p.E171*. By transfecting breast cancer cell lines, we observed increased growth and cell motility for all the investigated mutants compared to wild-type p27Kip1 transfected cells. Furthermore, we discovered that the mutant forms exhibited altered phosphorylation on key residues and different localization or degradation mechanisms in comparison to the wild-type protein and suggested a possible region as crucial for the lysosome-dependent degradation of the protein. Finally, the loss of p27Kip1 ability in blocking cell proliferation was in part explained through the different binding efficiency that mutant p27Kip1 forms exhibited with Cyclin/Cyclin-dependent Kinase complexes (or proteins involved indirectly in that binding) with respect to the WT.
A Pituitary Macroadenoma Cosecreting Prolactin and Growth Hormone in a Patient With Multiple Endocrine Neoplasia Type 4.
Multiple endocrine neoplasia type-4 (MEN4) is a rare form of multiple endocrine neoplasia due to a pathogenic variation in the cyclin-dependent kinase inhibitor 1B (CDKN1B) gene. It has a similar presentation to patients with multiple endocrine neoplasia type-1 (MEN1), with primary hyperparathyroidism and pituitary adenomas being the most common features. In this case, we describe a 54-year-old woman presenting with a pituitary macroadenoma cosecreting growth hormone and prolactin and primary hyperparathyroidism. She was initially managed with cabergoline without satisfactory response. Eventually she proceeded to transsphenoidal pituitary resection of the adenoma, and histology revealed appearances consistent with a mixed somatotroph-lactotroph adenoma. Subsequently genetic analysis confirmed the presence of a pathogenic variant in the CDKN1B gene (CDKN1B c.410del), in keeping with a diagnosis of MEN4. This is the first case of a cosecreting pituitary macroadenoma to be described in a patient with MEN4.
A novel likely pathogenic germline variant in CDKN1B in a patient with MEN4 and medullary thyroid cancer.
Multiple endocrine neoplasia type 4 (MEN4) is caused by a germline CDKN1B deleterious variant. CDKN1B encodes p27Kip1, a cyclin-dependent kinase inhibitor that acts as tumor-suppressor. Clinical presentation of MEN4 is similar to multiple endocrine neoplasia type 1 (MEN1) but the diagnosis of MEN4 can only be established once a germline CDKN1B pathogenic variant has been confirmed. We describe a unique case presenting with two -rare endocrine conditions. A 59-year-old female patient was diagnosed with medullary thyroid cancer (MTC) without evidence of a germline pathogenic variant in the RET proto-oncogene. Five years later, she developed Cushing's disease. A heterozygous germline variant was identified in the CDKN1B gene, specifically c.536del (p.Prol179GlnfsTer46), corresponding to a single-nucleotide deletion at position 536. This variant induces a frameshift, leading to an alternative stop codon. Immunostaining of the pituitary and thyroid tumors revealed a weak nuclear expression of p27/Kip1 without significant differences of expression between tumor and non-tumoral tissues. The NGS panel (Oncomine Comprehensive Assay v3) performed in both MTC and pituitary tissues identified the germline CDKN1B variant, as well as a pathogenic missense somatic variant c.182 A > G, p.(Gln61Arg) in HRAS in the MTC, without any RET somatic pathogenic variant. Evaluation of loss of heterozygosity (LOH) in both MTC and pituitary tissues showed compatibility with copy-neutral LOH, although further evidence is required for definitive confirmation. In conclusion, we report a clinical case of MTC coexisting with MEN4 due to a novel CDKN1B germline heterozygote frameshift variant.
Anaplastic meningioma in a 6-year-old with somatic YAP1::MAML2 fusion and multiple endocrine neoplasia type 4 (MEN4) syndrome.
Meningiomas are the most common primary brain tumors in adults but much less frequent in children. Many subtypes exist, including anaplastic (malignant) meningioma, which accounts for less than 20% of pediatric tumors. Meningiomas can arise in association with cancer predisposition syndromes due to germline variants in genes such as NF2, MEN1 and SMARCE1. This report describes a 6-year-old boy diagnosed with anaplastic meningioma who was treated with surgery and focal radiation therapy. The family consented to participate in the Texas KidsCanSeq clinical genomics study. Analysis of germline and tumor samples detected a single germline finding of a CDKN1B pathogenic frameshift variant associated with Multiple Endocrine Neoplasia Type 4 (MEN4) without somatic loss of the other allele. Tumor analysis revealed a YAP1::MAML2 fusion, which has been previously reported in pediatric meningiomas not associated with NF2. YAP1::MAML2 fusion is a known driver for development of meningioma, but the role of the germline CDKN1B variant in the absence of a tumor second hit is unclear. This case highlights the importance of performing combined tumor and germline molecular genetic analysis of rare tumors to help clarify the risk of development of cancer in patients with rare cancer predisposition syndromes.
Chapter 6: Syndromic primary hyperparathyroidism.
Syndromic primary hyperparathyroidism has several features in common: younger age at diagnosis when compared with sporadic primary hyperparathyroidism, often synchronous or metachronous multi-glandular involvement, higher possibility of recurrence, association with other endocrine or extra-endocrine disorders, and suggestive family background with autosomal dominant inheritance. Hyperparathyroidism in multiple endocrine neoplasia type 1 is the most common syndromic hyperparathyroidism. It is often asymptomatic in adolescents and young adults, but may be responsible for recurrent lithiasis and/or bone loss. Hyperparathyroidism-jaw tumor syndrome is less frequent, but often immediately symptomatic, with higher blood calcium levels, and is sometimes associated with an atypic parathyroid tumor or parathyroid carcinoma. Hyperparathyroidism in multiple endocrine neoplasia type 2A is not at the forefront of the clinical picture, rarely revealing the disease, and often manifests with few symptoms. Multiple endocrine neoplasia type 4 is a more recently described entity, in which hyperparathyroidism seems to occur later and be less severe than in previous syndromes. In all cases, the indications and modalities of surgical treatment should be discussed in an expert center. The risk of recurrence after surgery, particularly high in multiple endocrine neoplasia type 1, requires long-term monitoring.
Publicações recentes
Phenotypic characterization of a new case of multiple endocrine neoplasia type 4 associated with de novo germline and somatic CDKN1B pathogenic variants.
A Pituitary Macroadenoma Cosecreting Prolactin and Growth Hormone in a Patient With Multiple Endocrine Neoplasia Type 4.
A novel likely pathogenic germline variant in CDKN1B in a patient with MEN4 and medullary thyroid cancer.
Anaplastic meningioma in a 6-year-old with somatic YAP1::MAML2 fusion and multiple endocrine neoplasia type 4 (MEN4) syndrome.
p27(Kip1) and Tumors: Characterization of CDKN1B Variants Identified in MEN4 and Breast Cancer.
📚 EuropePMC2.789 artigos no totalmostrando 27
Phenotypic characterization of a new case of multiple endocrine neoplasia type 4 associated with de novo germline and somatic CDKN1B pathogenic variants.
Annales d'endocrinologieA Pituitary Macroadenoma Cosecreting Prolactin and Growth Hormone in a Patient With Multiple Endocrine Neoplasia Type 4.
JCEM case reportsA novel likely pathogenic germline variant in CDKN1B in a patient with MEN4 and medullary thyroid cancer.
Familial cancerAnaplastic meningioma in a 6-year-old with somatic YAP1::MAML2 fusion and multiple endocrine neoplasia type 4 (MEN4) syndrome.
Cancer geneticsp27Kip1 and Tumors: Characterization of CDKN1B Variants Identified in MEN4 and Breast Cancer.
CellsChapter 6: Syndromic primary hyperparathyroidism.
Annales d'endocrinologieBeyond MEN1, When to Think About MEN4? Retrospective Study on 5600 Patients in the French Population and Literature Review.
The Journal of clinical endocrinology and metabolismPancreatic Neuroendocrine Tumor in a Patient With Multiple Endocrine Neoplasia Type 4.
Mayo Clinic proceedingsGenetic testing for familial hyperparathyroidism: clinical-genetic profile in a Mediterranean cohort.
Frontiers in endocrinologyClinical Factors Predicting Multiple Endocrine Neoplasia Type 1 and Type 4 in Patients with Neuroendocrine Tumors.
GenesMultiple endocrine neoplasia type 4 (MEN4): a thorough update on the latest and least known men syndrome.
EndocrineMultiple endocrine neoplasia type 4: a new member of the MEN family.
Endocrine connectionsThe Spectrum of Familial Pituitary Neuroendocrine Tumors.
Endocrine pathologyNovel germline variants of CDKN1B and CDKN2C identified during screening for familial primary hyperparathyroidism.
Journal of endocrinological investigationMultiple endocrine neoplasia type 4 & primary hyperparathyroidism: What the surgeon needs to know.
American journal of surgeryCase Report: New CDKN1B Mutation in Multiple Endocrine Neoplasia Type 4 and Brief Literature Review on Clinical Management.
Frontiers in endocrinologyMEN4, the MEN1 Mimicker: A Case Series of three Phenotypically Heterogenous Patients With Unique CDKN1B Mutations.
The Journal of clinical endocrinology and metabolismPancreatic neuroendocrine neoplasms: Updates on genomic changes in inherited tumour syndromes and sporadic tumours based on WHO classification.
Critical reviews in oncology/hematologyInherited syndromes involving pancreatic neuroendocrine tumors.
Journal of gastrointestinal oncologyMultiple Endocrine Neoplasia Type 4: Novel CDNK1B variant and immune anomalies.
Annales d'endocrinologieClinical and Molecular Update on Genetic Causes of Pituitary Adenomas.
Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolismeGermline CDKN1B Loss-of-Function Variants Cause Pediatric Cushing's Disease With or Without an MEN4 Phenotype.
The Journal of clinical endocrinology and metabolismCo-occurrence of multiple endocrine neoplasia type 4 and spinal neurofibromatosis: a case report.
Familial cancer[Inherited tumor syndromes of gastroenteropancreatic and thoracic neuroendocrine neoplasms].
Annales de pathologieSomatic and germline mutations in the pathogenesis of pituitary adenomas.
European journal of endocrinologyClinical Features of Multiple Endocrine Neoplasia Type 4: Novel Pathogenic Variant and Review of Published Cases.
The Journal of clinical endocrinology and metabolismUncommon association of cerebral meningioma, parathyroid adenoma and papillary thyroid carcinoma in a patient harbouring a rare germline variant in the CDKN1B gene.
BMJ case reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Neoplasia endócrina múltipla tipo 4.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Neoplasia endócrina múltipla tipo 4
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- p27Kip1 and Tumors: Characterization of CDKN1B Variants Identified in MEN4 and Breast Cancer.
- A Pituitary Macroadenoma Cosecreting Prolactin and Growth Hormone in a Patient With Multiple Endocrine Neoplasia Type 4.
- A novel likely pathogenic germline variant in CDKN1B in a patient with MEN4 and medullary thyroid cancer.
- Anaplastic meningioma in a 6-year-old with somatic YAP1::MAML2 fusion and multiple endocrine neoplasia type 4 (MEN4) syndrome.
- Chapter 6: Syndromic primary hyperparathyroidism.
- Phenotypic characterization of a new case of multiple endocrine neoplasia type 4 associated with de novo germline and somatic CDKN1B pathogenic variants.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:276152(Orphanet)
- OMIM OMIM:610755(OMIM)
- MONDO:0012552(MONDO)
- GARD:17275(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q26492830(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
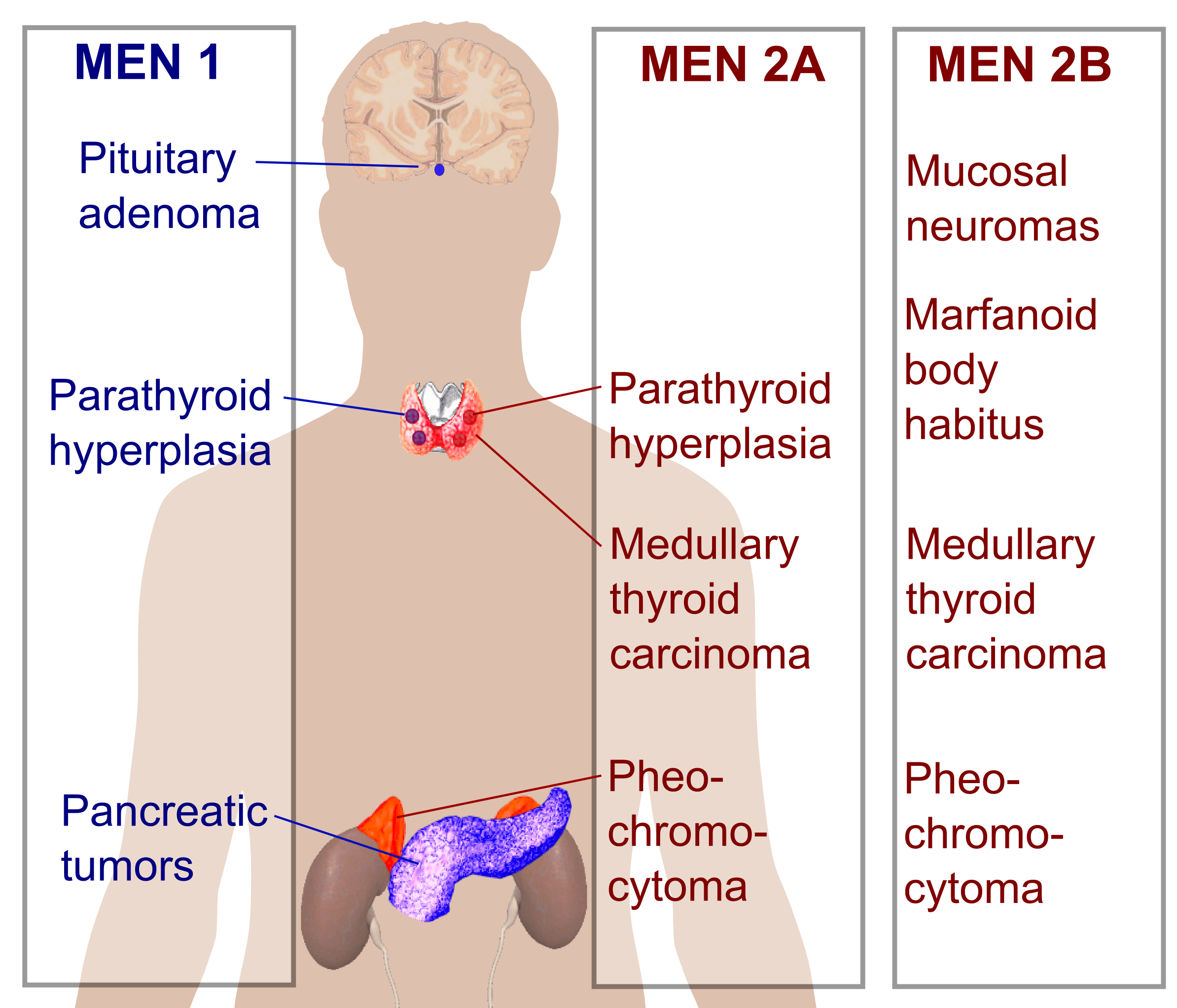
Neoplasia endócrina múltipla tipo 4
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos aprovados FDA
- fonte: FDA OpenFDA
- Ensaios clínicos
- fonte: ClinicalTrials.gov