A osteogênese imperfeita tipo I é um tipo leve de osteogênese imperfeita (OI), uma doença genética caracterizada por aumento da fragilidade óssea, baixa massa óssea e suscetibilidade a fraturas ósseas.
Introdução
O que você precisa saber de cara
A osteogênese imperfeita tipo I é um tipo leve de osteogênese imperfeita (OI), uma doença genética caracterizada por aumento da fragilidade óssea, baixa massa óssea e suscetibilidade a fraturas ósseas.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 5 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 21 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
5 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisThis multifunctional protein catalyzes the formation, breakage and rearrangement of disulfide bonds. At the cell surface, seems to act as a reductase that cleaves disulfide bonds of proteins attached to the cell. May therefore cause structural modifications of exofacial proteins. Inside the cell, seems to form/rearrange disulfide bonds of nascent proteins. At high concentrations and following phosphorylation by FAM20C, functions as a chaperone that inhibits aggregation of misfolded proteins (Pub
Endoplasmic reticulumEndoplasmic reticulum lumenMelanosomeCell membrane
Cole-Carpenter syndrome 1
A form of Cole-Carpenter syndrome, a disorder characterized by features of osteogenesis imperfecta such as bone deformities and severe bone fragility with frequent fractures, in association with craniosynostosis, ocular proptosis, hydrocephalus, growth failure and distinctive facial features. Craniofacial findings include marked frontal bossing, midface hypoplasia, and micrognathia. Despite the craniosynostosis and hydrocephalus, intellectual development is normal. CLCRP1 inheritance is autosomal dominant.
Zinc metalloprotease that mediates intramembrane proteolysis of proteins such as ATF6, ATF6B, SREBF1/SREBP1 and SREBF2/SREBP2 (PubMed:10805775, PubMed:11163209). Catalyzes the second step in the proteolytic activation of the sterol regulatory element-binding proteins (SREBPs) SREBF1/SREBP1 and SREBF2/SREBP2: cleaves SREBPs within the first transmembrane segment, thereby releasing the N-terminal segment with a portion of the transmembrane segment attached (PubMed:10805775, PubMed:27380894, PubMed
MembraneCytoplasmGolgi apparatus membrane
IFAP syndrome 1, with or without Bresheck syndrome
An X-linked syndrome characterized by a peculiar triad of follicular ichthyosis, total or subtotal atrichia, and photophobia of varying degree. Histopathologically, the epidermal granular layer is generally well-preserved or thickened at the infundibulum. Hair follicles are poorly developed and tend to be surrounded by an inflammatory infiltrate. A subgroup of patients is described with lamellar rather than follicular ichthyosis. Non-consistent features may include growth and psychomotor retardation, aganglionic megacolon, seizures and nail dystrophy.
Type I collagen is a member of group I collagen (fibrillar forming collagen)
Secreted, extracellular space, extracellular matrix
Ehlers-Danlos syndrome, arthrochalasia type, 2
A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSARTH2 is an autosomal dominant condition characterized by frequent congenital hip dislocation and extreme joint laxity with recurrent joint subluxations and minimal skin involvement.
Type I collagen is a member of group I collagen (fibrillar forming collagen)
Secreted, extracellular space, extracellular matrix
Caffey disease
An autosomal dominant disorder characterized by an infantile episode of massive subperiosteal new bone formation that typically involves the diaphyses of the long bones, mandible, and clavicles. The involved bones may also appear inflamed, with painful swelling and systemic fever often accompanying the illness. The bone changes usually begin before 5 months of age and resolve before 2 years of age.
Component of the coat protein complex II (COPII) which promotes the formation of transport vesicles from the endoplasmic reticulum (ER). The coat has two main functions, the physical deformation of the endoplasmic reticulum membrane into vesicles and the selection of cargo molecules for their transport to the Golgi complex (PubMed:17499046, PubMed:18843296, PubMed:20427317). Plays a central role in cargo selection within the COPII complex and together with SEC24C may have a different specificity
Cytoplasmic vesicle, COPII-coated vesicle membraneEndoplasmic reticulum membraneCytoplasm, cytosol
Cole-Carpenter syndrome 2
A form of Cole-Carpenter syndrome, a disorder characterized by features of osteogenesis imperfecta such as bone deformities and severe bone fragility with frequent fractures, in association with craniosynostosis, ocular proptosis, hydrocephalus, growth failure and distinctive facial features. Craniofacial findings include marked frontal bossing, midface hypoplasia, and micrognathia. Despite the craniosynostosis and hydrocephalus, intellectual development is normal. CLCRP2 inheritance is autosomal recessive.
Variantes genéticas (ClinVar)
2.857 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 7.473 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
49 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Osteogenesis imperfecta tipo 1
Centros de Referência SUS
24 centros habilitados pelo SUS para Osteogenesis imperfecta tipo 1
Centros para Osteogenesis imperfecta tipo 1
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
3 ensaios clínicos encontrados, 2 ativos.
Publicações mais relevantes
Serum Osteocalcin in Pediatric Osteogenesis Imperfecta: Impact of Disease Type and Bisphosphonate Therapy.
The aim of this study was to analyze the factors that may influence serum osteocalcin levels in children with osteogenesis imperfecta treated with intravenous sodium pamidronate and to define the role of osteocalcin assessment. The study included 61 patients diagnosed with osteogenesis imperfecta type 1 or 3, aged 2 to 18, hospitalized for intravenous sodium pamidronate administration. A retrospective analysis of medical records was conducted, collecting information on age, sex, body weight, height, the number of long bone fractures throughout life, serum levels of osteocalcin, creatinine, alkaline phosphatase, 25(OH)D3, and DXA BMD z-scores for the L1-L4 spine segment. The concentration of osteocalcin is higher in patients with osteogenesis imperfecta than the reference ranges for sex and age. Patients diagnosed with type 3 have significantly lower osteocalcin levels compared to patients with type 1. Also, increasing the age-standardized pamidronate cycle rate significantly reduced osteocalcin concentration. The strongest predictor of osteocalcin concentration among the factors studied is the type of osteogenesis imperfecta. L1-L4 BMD value and fracture frequency were unrelated to osteocalcin concentration. Osteocalcin is an important marker of bone formation that should be measured at the beginning of treatment, as its concentration decreases after successive doses of bisphosphonates.
New Lens On Congenital Mild Bone Fragility: a Novel Col1a1 Knockout Mouse Model for Osteogenesis Imperfecta Type 1.
Osteogenesis imperfecta (OI) is a genetic disorder characterized by bone fragility. It is one of the most prevalent rare skeletal dysplasias. The mildest form, OI type 1, predominantly results from collagen type I haploinsufficiency due to pathogenic variants in the COL1A1 gene, leading to reduced collagen type I. Despite OI type 1 representing approximately half of the OI population, the lack of an effective mouse model has hindered research and therapy development(1). To address this gap, we developed a genetically engineered mouse model harbouring a heterozygous deletion of the Col1a1 allele using the CRISPR/Cas system. The bone phenotype was characterised in 8- and 24-week-old mice, assessing transcriptomics and serum markers for bone formation (procollagen type I N-terminal propeptide) and resorption (tartrate-resistant acid phosphatase 5b). Bone volume, microarchitecture, and strength were evaluated by micro-computed tomography, histomorphometry and three-point bending test. We showed that the decreased Col1a1 to Col1a2 mRNA ratio determines reduced collagen type I production in OI mice bones as the underlying mechanism of haploinsufficient OI. This was supported by COL1A1 to COL1A2 mRNA ratio findings in human OI cell models, including fibroblasts and induced mesenchymal stem cells, as well as in induced pluripotent and mesenchymal stem cell models that were edited to carry a heterozygous COL1A1 allele. Our findings indicate for the first time that reduced bone volume and altered bone microarchitecture in haploinsufficient OI depends on the Col1a1 to Col1a2 mRNA ratio regulation. This novel mouse model faithfully recapitulates OI type 1 and provides a vital tool for investigating the disease mechanism and developing targeted therapeutic strategies for this large neglected OI patient population. Osteogenesis imperfecta is the most common genetic disease of bone fragility. In half of these patients, it is caused by less collagen production, which makes bones fragile. Surprisingly, there is still no reliable mouse model for this large patient group, which has hindered disease understanding and therapy development. In our study, we engineered a successful mouse model with collagen deficiency, which faithfully mimics the clinical and molecular disease properties. Together with findings in patient bone cells and collagen gene-edited bone cell models, we also present new insights into the underlying collagen-based OI mechanism in this large, neglected patient group.
Osteogenesis Imperfecta type 1: like mother, like daughter - Challenges in the perinatal management.
A third gravida with osteogenesis imperfecta (OI) type 1, in her 20s, was referred from the Medical Genetics department at 12+ weeks with a prenatal diagnosis of OI type 1 in this fetus for further management. She was wheelchair-bound and keen to continue this pregnancy. She had medical termination in her two previous pregnancies for OI in the fetuses. Ultrasound at 12+ weeks revealed a short-bent femur with sparing of the long bones of the upper limb. Serial ultrasound revealed progressive affliction of the long bones with falling growth profile and polyhydramnios. She was delivered at 36 weeks by caesarean for breech in labour under regional anaesthesia.A multidisciplinary approach, patient determination, and good partner support helped in the successful management of this pregnancy.The neonate had blue sclera, dentigerous imperfecta, bowing of the femur and relatively spared upper limbs. Growth was on the third centile. The mother says she brings the girl for follow-up every 3-6 months to give injection zoledronate. The mother confirms her girl can stand with support, crawl, and speak two-syllable words. Her daughter had to undergo femur corrective osteotomy rush nailing and hip spice application for a closed fracture of the left femur.
The Natural History of Symptomatic Fractures in Children and Adolescents with Osteogenesis Imperfecta Type 1: A Cohort Study from Western Australia.
The fracture experience of children and adolescents with osteogenesis imperfecta (OI) type 1 is not well described in the literature. We present data on symptomatic long bones and axial skeleton fractures of all patients aged 0 to 18 years with OI type 1 seen at a specialized bone clinic in Western Australia in the period 2008 to 2020 using a retrospective chart review method. The cohort consisted of 44 patients (21 males, 23 females). Median (interquartile range [IQR]) age was 11.3 (6.2 to 17) years, giving a total of 520 patient-years in the study during which 197 fractures were experienced. The mean fracture rate was 379 fractures per 1000 patient-years (95% confidence interval [CI]: 310 to 440); however, the experience for fractures varied from ≤1 fracture in 23% (n = 10) to two to 20 in 77% (n = 34) of the cohort. Twenty-one patients (48.5%) received bisphosphonates during the study period. In logistic regression, age, but not sex or family history of OI, was a significant predictor of fracture risk. The highest total fracture rate was observed in the age group 0 to <3 years at 469 fractures/1000 patient-years, which declined to 140 fractures/1000 patient-years in the age group 15 to 18 years. The lower limbs were the site of 49.7% of all fractures. The highest rate for lower limb fracture was in the age group 0 to <3 years at 331 fractures/1000 patient-years, decreasing to 0 fractures/1000 patient-years in the age group 15 to 18 years. Upper limb fracture rates increased from 100 fractures/1000 patient-years in the 0 to <3 years age group to 307 fractures/1000 patient-years in the 9 to <12 years age group and then declining to 70 fractures/1000 years in the 15 to 18 years age group. In pediatric patients with OI type 1, fracture risk is highest in early life, especially in the lower limbs. Multidisciplinary care of children with OI should have a particular focus on strategies to prevent these fractures. © 2023 The Authors. JBMR Plus published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research.
Exploration of the skeletal phenotype of the Col1a1 +/Mov13 mouse model for haploinsufficient osteogenesis imperfecta type 1.
Osteogenesis Imperfecta is a rare genetic connective tissue disorder, characterized by skeletal dysplasia and fragile bones. Currently only two mouse models have been reported for haploinsufficient (HI) mild Osteogenesis Imperfecta (OI); the Col1a1 +/Mov13 (Mov13) and the Col1a1 +/-365 mouse model. The Mov13 mice were created by random insertion of the Mouse Moloney leukemia virus in the first intron of the Col1a1 gene, preventing the initiation of transcription. Since the development of the Mov13 mice almost four decades ago and its basic phenotypic characterization in the 90s, there have not been many further studies. We aimed to extensively characterize the Mov13 mouse model in order to critically evaluate its possible use for preclinical studies of HI OI. Bone tissue from ten heterozygous Mov13 and ten wild-type littermates (WT) C57BL/6J mice (50% males per group) was analyzed at eight weeks of age with bone histomorphometry, micro computed tomography (microCT), 3-point bending, gene expression of different collagens, as well as serum markers of bone turnover. The Mov13 mouse presented a lower bone strength and impaired material properties based on our results of 3-point bending and microCT analysis respectively. In contrast, no significant differences were found for all histomorphometric parameters. In addition, no significant differences in Col1a1 bone expression were present, but there was a significant lower P1NP concentration, a bone formation marker, measured in serum. Furthermore, bone tissue of Mov13 mice presented significantly higher expression of collagens (Col1a2, Col5a1 and Col5a2), and bone metabolism markers (Bglap, Fgf23, Smad7, Edn1 and Eln) compared to WT. Finally, we measured a significantly lower Col1a1 expression in heart and skin tissue and also determined a higher expression of other collagens in the heart tissue. Although we did not detect a significant reduction in Col1a1 expression in the bone tissue, a change in bone structure and reduction in bone strength was noted. Regrettably, the variability of the bone phenotype and the appearance of severe lymphoma in adult Mov13 mice, does not favor their use for the testing of new long-term drug studies. As such, a new HI OI type 1 mouse model is urgently needed.
Publicações recentes
New Lens On Congenital Mild Bone Fragility: a Novel Col1a1 Knockout Mouse Model for Osteogenesis Imperfecta Type 1.
Serum Osteocalcin in Pediatric Osteogenesis Imperfecta: Impact of Disease Type and Bisphosphonate Therapy.
Osteogenesis Imperfecta type 1: like mother, like daughter - Challenges in the perinatal management.
The Natural History of Symptomatic Fractures in Children and Adolescents with Osteogenesis Imperfecta Type 1: A Cohort Study from Western Australia.
Exploration of the skeletal phenotype of the Col1a1 (+/Mov13) mouse model for haploinsufficient osteogenesis imperfecta type 1.
📚 EuropePMC4.738 artigos no totalmostrando 15
New Lens On Congenital Mild Bone Fragility: a Novel Col1a1 Knockout Mouse Model for Osteogenesis Imperfecta Type 1.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchSerum Osteocalcin in Pediatric Osteogenesis Imperfecta: Impact of Disease Type and Bisphosphonate Therapy.
International journal of molecular sciencesOsteogenesis Imperfecta type 1: like mother, like daughter - Challenges in the perinatal management.
BMJ case reportsThe Natural History of Symptomatic Fractures in Children and Adolescents with Osteogenesis Imperfecta Type 1: A Cohort Study from Western Australia.
JBMR plusExploration of the skeletal phenotype of the Col1a1 +/Mov13 mouse model for haploinsufficient osteogenesis imperfecta type 1.
Frontiers in endocrinologyBrain-Type Creatine Kinase Release from Cultured Osteoclasts Exposed to Neridronate in Children Affected by Osteogenesis Imperfecta Type 1.
BiomedicinesAccelerated mineralization kinetics in children with osteogenesis imperfecta type 1.
Bone[The coincidence of benign non-familial infantile seizures type 2 with osteogenesis imperfecta type 1].
Zhurnal nevrologii i psikhiatrii imeni S.S. KorsakovaBilateral Asynchronous Displaced Olecranon Fractures in a Patient With Osteogenesis Imperfecta.
CureusFunctional outcomes of an adult with osteogenesis imperfecta after rehabilitation post bilateral Girdlestone procedure.
BMJ case reportsEhlers-Danlos Syndrome: Immunologic contrasts and connective tissue comparisons.
Journal of translational autoimmunityOsteogenesis imperfecta type 1 with an incidental finding of bilateral radioulnar synostosis.
Clinical dysmorphologyOsteogenesis Imperfecta Type 1: When Eyes Talk About Bones.
Journal of clinical rheumatology : practical reports on rheumatic & musculoskeletal diseasesEarly Motor Delay: An Outstanding, Initial Sign of Osteogenesis Imperfecta Type 1.
Journal of orthopaedic case reportsFamily experience with osteogenesis imperfecta type 1: the most distressing situations.
Disability and rehabilitationAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Osteogenesis imperfecta tipo 1.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Osteogenesis imperfecta tipo 1
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Serum Osteocalcin in Pediatric Osteogenesis Imperfecta: Impact of Disease Type and Bisphosphonate Therapy.
- New Lens On Congenital Mild Bone Fragility: a Novel Col1a1 Knockout Mouse Model for Osteogenesis Imperfecta Type 1.Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research· 2025· PMID 41051363mais citado
- Osteogenesis Imperfecta type 1: like mother, like daughter - Challenges in the perinatal management.
- The Natural History of Symptomatic Fractures in Children and Adolescents with Osteogenesis Imperfecta Type 1: A Cohort Study from Western Australia.
- Exploration of the skeletal phenotype of the Col1a1 +/Mov13 mouse model for haploinsufficient osteogenesis imperfecta type 1.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:216796(Orphanet)
- OMIM OMIM:166200(OMIM)
- MONDO:0008146(MONDO)
- GARD:8694(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q27677735(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
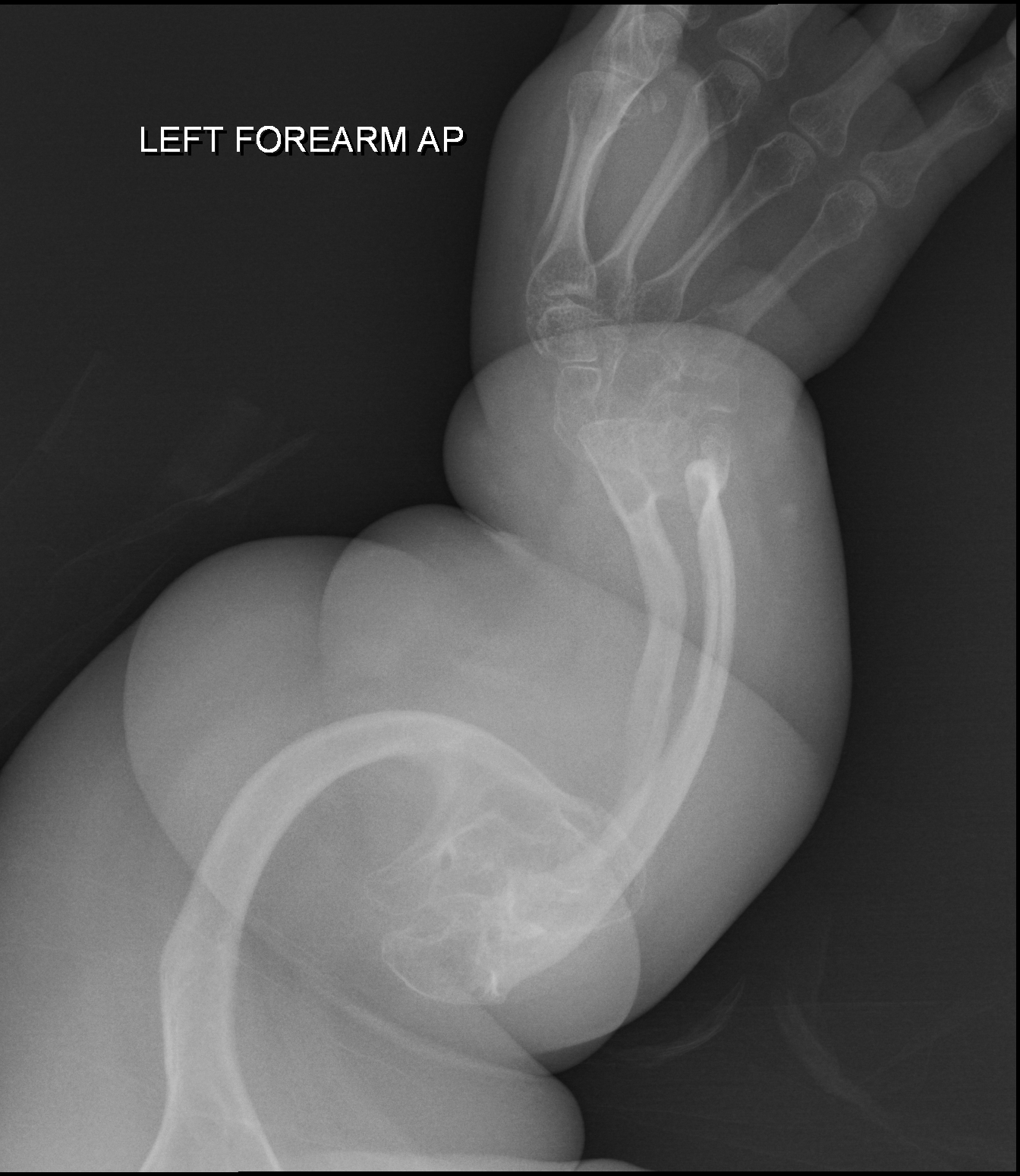
Osteogenesis imperfecta tipo 1
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov