A síndrome da córnea frágil é uma forma de síndrome de Ehlers-Danlos caracterizada por manifestações oculares graves devido ao extremo adelgaçamento e fragilidade da córnea com ruptura na ausência de trauma significativo e progressão para cegueira. As manifestações extraoculares incluem surdez, displasia do desenvolvimento do quadril e hipermobilidade articular.
Introdução
O que você precisa saber de cara
A síndrome da córnea frágil é uma forma de síndrome de Ehlers-Danlos caracterizada por manifestações oculares graves devido ao extremo adelgaçamento e fragilidade da córnea com ruptura na ausência de trauma significativo e progressão para cegueira. As manifestações extraoculares incluem surdez, displasia do desenvolvimento do quadril e hipermobilidade articular.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 15 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 55 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
May be involved in transcriptional regulation
Nucleus
Brittle cornea syndrome 1
An autosomal recessive disorder characterized by extreme corneal thinning resulting in corneal rupture after minor trauma, blue sclerae, keratoconus or keratoglobus, hyperelasticity of the skin, and hypermobility of the joints. It shares some features with, but is much less severe than, the ocular form of Ehlers-Danlos syndrome (EDS6).
Sequence-specific DNA-binding transcription factor. Represses transcription at least in part by recruitment of the histone methyltransferase EHMT2/G9A and histone deacetylases such as HDAC1. Regulates hematopoiesis-associated protein-coding and microRNA (miRNA) genes. May regulate the expression of proteins involved in extracellular matrix development and maintenance, including fibrillar collagens, such as COL4A1 and COL11A1, connective tissue components, such as HAPLN1, and molecules regulating
Nucleus
Brittle cornea syndrome 2
A disorder characterized by extreme corneal thinning resulting in corneal rupture after minor trauma, blue sclerae, keratoconus or keratoglobus, hyperelasticity of the skin, and hypermobile joints.
Variantes genéticas (ClinVar)
940 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 285 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome da córnea frágil
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
1 ensaios clínicos encontrados.
Publicações mais relevantes
The Genetic and Epigenetic Architecture of Keratoconus: Emerging Pathways and Clinical Implications.
Keratoconus (KC) is a progressive corneal ectasia and a leading cause of corneal transplantation in young adults. Once regarded as a biomechanical disorder, KC is now recognized as a complex disease driven by genetic predisposition, epigenetic modulation, and environmental triggers. Advances in genomics and transcriptomics have begun to elucidate the molecular mechanisms underlying corneal thinning and ectasia. This review synthesizes two decades of evidence on the genetic and epigenetic architecture of keratoconus, highlights key molecular pathways implicated by these findings, and discusses translational implications for early diagnosis, risk prediction, and novel therapeutic strategies. A narrative review was conducted of peer-reviewed human, animal, and in vitro studies published from 2000 to 2025, with emphasis on genome-wide association studies (GWAS), sequencing data, methylation profiling, and non-coding RNA analyses. Findings were integrated with functional studies linking genetic variation to molecular and biomechanical phenotypes. Genetic studies consistently implicate loci such as ZNF469, COL5A1, LOX, HGF, FOXO1, and WNT10A, alongside rare variants in Mendelian syndromes (e.g., brittle cornea syndrome, Ehlers-Danlos spectrum). Epigenetic research demonstrates altered DNA methylation, dysregulated microRNAs (e.g., MIR184, miR-143, miR-182), and aberrant lncRNA networks influencing extracellular matrix remodeling, collagen cross-linking, oxidative stress, and inflammatory signaling. Gene-environment interactions, particularly with eye rubbing and atopy, further shape disease expression. Translational progress includes polygenic risk scores, tear-based biomarkers, and early preclinical studies using RNA-based approaches (including siRNA and antisense oligonucleotides targeting matrix-degrading and profibrotic pathways) and proof-of-concept gene-editing strategies demonstrated in corneal cell and ex vivo models. Conclusions: Keratoconus arises from the convergence of inherited genomic risk, epigenetic dysregulation, and environmental stressors. Integrating multi-omic insights into clinical practice holds promise for earlier detection, precision risk stratification, and development of targeted therapies that move beyond biomechanical stabilization to disease modification.
Brittle cornea syndrome: Integrating unique presentations and novel management options.
Brittle cornea syndrome (BCS) is a connective tissue disorder with a vast majority of clinical presentations and prognostic outcomes. This case series highlights various ocular presentations, their responses to treatment strategies, and final visual outcomes. It was an observational case series at a tertiary eye care center. Three patients with BCS and different corneal pathologies were managed and studied. The first case presented with wound dehiscence, a shallow anterior chamber, and a disenclaved iris claw lens. He underwent intraocular lens (IOL) explantation and wound closure using cyanoacrylate glue and a bandage contact lens. The second case involved corneal ectasia with rupture and an intumescent cataract, along with lens-endothelial touch. He underwent lens aspiration with posterior chamber IOL implantation and epi-keratoplasty with a biosynthetic cornea. However, the biosynthetic cornea had to be removed due to a severe inflammatory reaction following the procedure. The patient was later rehabilitated with deep anterior lamellar keratoplasty. The third patient presented with bilateral tractional descemet membrane detachment resulting from multiple minor injuries. Refraction with prescription glasses was used for visual rehabilitation. In conclusion, the management of BCS requires individualized treatment strategies tailored to each patient's presentation and response.
Brittle Cornea Syndrome Type 1 in Siblings: Severe Presentation in One and Misdiagnosis as Primary Congenital Glaucoma in the Other.
The purpose of this study was to present two cases of brittle cornea syndrome (BCS) in siblings, one with severe corneal perforation and the other misdiagnosed as primary congenital glaucoma, emphasizing clinical, histopathological, and genetic findings. This was a case report involving a 4-year-old boy and his 8-year-old brother, both presenting with thin, steep corneas. Clinical examination, Pentacam imaging, genetic analysis, systemic evaluation, and corneal histopathology were performed. The younger sibling presented with corneal perforation after trauma, and histopathology revealed a markedly thinned corneal stroma with disrupted collagen architecture. Genetic testing confirmed a zinc finger protein (ZNF)469 mutation, consistent with brittle cornea syndrome type 1 (BCS1). Sanger sequencing of the older sibling, who was previously misdiagnosed with primary congenital glaucoma, identified the same ZNF469 mutation. He exhibited corneal thinning, bluish sclera, and healthy optic discs. These cases highlight the variability in BCS presentations, the risk of misdiagnosis as glaucoma, and the importance of early genetic testing and protective measures. Histopathological findings provide additional insights into the structural abnormalities in BCS corneas.
Brittle Cornea Syndrome: Molecular Diagnosis and Management.
Background and Clinical Significance: Brittle cornea syndrome (BCS) is a rare, autosomal recessive connective tissue disorder characterized by extreme corneal thinning, high myopia, and increased risk of spontaneous or trauma-induced ocular rupture. It is primarily caused by mutations in the ZNF469 or PRDM5 genes, which regulate extracellular matrix integrity. Early recognition and diagnosis of BCS are crucial to prevent severe visual impairment. This report presents two genetically confirmed cases of BCS in Albanian siblings, emphasizing the diagnostic value of whole-exome sequencing and individualized surgical management strategies. Case Presentation: Two siblings-a 28-year-old male and a 25-year-old female-presented with progressive visual deterioration and marked corneal thinning (<200 µm). Both had a history of spontaneous ocular rupture following minor trauma in the contralateral eye. Detailed ophthalmologic evaluation revealed keratoglobus, high myopia, and irregular astigmatism. Genetic testing identified the homozygous pathogenic variant c.974delG (p.Cys325LeufsX2) in the PRDM5 gene in both cases. The male underwent penetrating keratoplasty (PKP), achieving a best-corrected visual acuity (BCVA) of 20/30. The female initially underwent deep anterior lamellar keratoplasty (DALK), which was converted to PKP intraoperatively due to central endothelial perforation, resulting in a BCVA of 20/25. Both patients remained complication-free over a 7-year follow-up period. Conclusions: These cases highlight the importance of early genetic diagnosis and a tailored surgical approach in managing BCS. Long-term monitoring and protective strategies are essential to prevent complications. Incorporating genetic testing into clinical practice can enhance diagnostic accuracy and guide personalized treatment plans in patients with hereditary corneal dystrophies.
[Ocular manifestations and genetic aspects of Ehlers-Danlos syndrome].
Ehlers-Danlos syndrome (EDS) is a heterogeneous group of inherited connective tissue diseases characterized by abnormal collagen synthesis and affecting various organs, including the eyes. This review analyses the current data on this disease, focusing on EDS types associated with ophthalmological manifestations: brittle cornea syndrome, kyphoscoliotic, musculocontractural, spondylodysplastic, dermatosparaxis, vascular, and classical types. The article describes ophthalmological diagnostic criteria for the different types of EDS and lists other possible ocular symptoms. The review also emphasizes the need for molecular genetic testing for accurate diagnosis in view of the difficulty in identifying specific genes encoding collagen or collagen interacting proteins, highlights the importance of timely treatment and describes methods of correcting visual disturbances, which are important for preventing severe complications such as globe rupture and retinal detachment. Синдром Элерса—Данлоса (EDS) представляет собой гетерогенную группу наследственных заболеваний соединительной ткани, характеризующихся аномальным синтезом коллагена и поражающих различные органы, включая глаза. В настоящем обзоре проанализированы современные данные об этом заболевании, с акцентом на типы EDS, ассоциированные с офтальмологическими проявлениями: синдром хрупкой роговицы, кифосколиотический, мышечноконтрактурный, спондилодиспластический, дерматоспараксисный, сосудистый и классический типы. Описаны офтальмологические диагностические критерии для различных типов синдрома Элерса—Данлоса, а также перечислены другие возможные глазные симптомы. Учитывая сложность идентификации конкретных генов, кодирующих коллаген или взаимодействующие с ним белки, подчеркнута необходимость молекулярно-генетического тестирования для точной диагностики. Выделена значимость своевременного лечения и описаны методы коррекции нарушений со стороны органа зрения, которые важны для предотвращения таких серьезных последствий, как разрывы глазного яблока и отслойка сетчатки.
Publicações recentes
Insight into genes responsible for cornea plana, megalocornea, keratoconus and brittle cornea syndrome.
Clinical and Molecular Features of 11 Patients with Different Subtypes of Ehlers-Danlos Syndrome.
The Genetic and Epigenetic Architecture of Keratoconus: Emerging Pathways and Clinical Implications.
Brittle cornea syndrome: Integrating unique presentations and novel management options.
Brittle Cornea Syndrome Type 1 in Siblings: Severe Presentation in One and Misdiagnosis as Primary Congenital Glaucoma in the Other.
📚 EuropePMC59 artigos no totalmostrando 55
The Genetic and Epigenetic Architecture of Keratoconus: Emerging Pathways and Clinical Implications.
GenesBrittle cornea syndrome: Integrating unique presentations and novel management options.
Oman journal of ophthalmologyBrittle Cornea Syndrome Type 1 in Siblings: Severe Presentation in One and Misdiagnosis as Primary Congenital Glaucoma in the Other.
CorneaBrittle Cornea Syndrome: Molecular Diagnosis and Management.
Diagnostics (Basel, Switzerland)[Ocular manifestations and genetic aspects of Ehlers-Danlos syndrome].
Vestnik oftalmologiiOn subcellular distribution of the zinc finger 469 protein (ZNF469) and observed discrepancy in the localization of endogenous and overexpressed ZNF469.
FEBS open bioA Possible Phenotype-to-Genotype Association of Novel Single-Nucleotide Variants in the Coding Exons of the ZNF469 Gene to Arterial Aneurysmal and Dissection Diseases.
International journal of molecular sciencesCongenital glaucoma in brittle cornea syndrome type 2 with a novel mutation in PRDM5.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusCataract surgery complications in a patient with brittle cornea syndrome and Ehlers-Danlos syndrome type six: A case report.
American journal of ophthalmology case reportsBrittle cornea syndrome: A novel mutation.
HeliyonAn Eye into the Aorta: The Role of Extracellular Matrix Regulatory Genes ZNF469 and PRDM5, from Their Previous Association with Brittle Cornea Syndrome to Their Novel Association with Aortic and Arterial Aneurysmal Diseases.
International journal of molecular sciencesVariants in the ZNF469 gene in families with Brittle cornea syndrome and keratoconus.
HeliyonBiallelic novel variants in ZNF469 causing Brittle Cornea Syndrome 1: a detailed report of an Indian patient.
Ophthalmic geneticsIdentification and Management of a Novel PRDM5 Gene Pathologic Variant in a Family With Brittle Cornea Syndrome.
CorneaCharacteristics of brittle cornea syndrome by multimodal imaging modalities: a case report.
BMC ophthalmologyZnf469 Plays a Critical Role in Regulating Synthesis of ECM: A Zebrafish Model of Brittle Cornea Syndrome.
Investigative ophthalmology & visual scienceNew ZNF469 Mutations in Spanish Siblings With Brittle Cornea Syndrome.
CorneaPenetrating keratoplasty in brittle Cornea syndrome: Case series and review of the literature.
European journal of ophthalmologyHomozygous Val6Gly Variation in PRDM5 Gene Causing Brittle Cornea Syndrome: A New Turkish Case.
Molecular syndromologyA novel homozygous ZNF469 variant causing brittle cornea syndrome is associated with corneal ectasias in heterozygous carriers.
International ophthalmologySpontaneous Rupture of an Infant's Cornea.
Journal of pediatric ophthalmology and strabismusBrittle cornea syndrome: A tale of three brothers.
Indian journal of ophthalmologyDoes brittle cornea syndrome have a bone fragility phenotype?
Bone reportsSurgical technique for the management of corneal perforation in brittle cornea.
Indian journal of ophthalmologyA mouse model of brittle cornea syndrome caused by mutation in Zfp469.
Disease models & mechanismsIdentification of the novel SDR42E1 gene that affects steroid biosynthesis associated with the oculocutaneous genital syndrome.
Experimental eye researchCorneal ectasia associated with posterior lamellar opacification.
Ophthalmic geneticsMore than meets the eye: Expanding and reviewing the clinical and mutational spectrum of brittle cornea syndrome.
Human mutationCorneal rupture management with amniotic membrane graft in a patient with Brittle cornea syndrome.
Journal francais d'ophtalmologieEhlers-Danlos Syndrome: Immunologic contrasts and connective tissue comparisons.
Journal of translational autoimmunityCase report of a PRDM5 linked brittle cornea syndrome type 2 in association with a novel SLC6A5 mutation.
Indian journal of ophthalmologyWhole-Exome Sequencing Identifies Novel Compound Heterozygous ZNF469 Mutations in Two Siblings with Mild Brittle Cornea Syndrome.
Calcified tissue internationalCase Series of Brittle Cornea Syndrome.
Case reports in ophthalmological medicineSystematic review of differential methylation in rare ophthalmic diseases.
BMJ open ophthalmologyBrittle cornea syndrome: current perspectives [Response to Letter].
Clinical ophthalmology (Auckland, N.Z.)Brittle cornea syndrome: current perspectives [Letter].
Clinical ophthalmology (Auckland, N.Z.)Brittle cornea syndrome: current perspectives.
Clinical ophthalmology (Auckland, N.Z.)Corneal Perforation After Corneal Cross-Linking in Keratoconus Associated With Potentially Pathogenic ZNF469 Mutations.
CorneaBrittle cornea syndrome: Disease-causing mutations in ZNF469 and two novel variants identified in a patient followed for 26 years.
Biomedical papers of the Medical Faculty of the University Palacky, Olomouc, CzechoslovakiaIdentification of a Novel ZNF469 Mutation in a Pakistani Family With Brittle Cornea Syndrome.
Cornea[Brittle cornea syndrome type 1 caused by compound heterozygosity of two mutations in the ZNF469 gene].
Der Ophthalmologe : Zeitschrift der Deutschen Ophthalmologischen GesellschaftBrittle cornea syndrome: a case report and review of the literature.
BMC ophthalmologyUse of an onlay corneal lamellar graft for brittle cornea syndrome.
BMJ case reportsRare, Potentially Pathogenic Variants in ZNF469 Are Not Enriched in Keratoconus in a Large Australian Cohort of European Descent.
Investigative ophthalmology & visual scienceGenetic factors influencing the reduction of central corneal thickness in disorders affecting the eye.
Ophthalmic geneticsUnusual case of globe perforation: the brittle cornea without systemic manifestations.
BMJ case reportsGenetics in Keratoconus: where are we?
Eye and vision (London, England)Identification of Mutations in the PRDM5 Gene in Brittle Cornea Syndrome.
CorneaBruch's membrane abnormalities in PRDM5-related brittle cornea syndrome.
Orphanet journal of rare diseasesWhole exome sequencing identifies a heterozygous missense variant in the PRDM5 gene in a family with Axenfeld-Rieger syndrome.
NeurogeneticsA role for repressive complexes and H3K9 di-methylation in PRDM5-associated brittle cornea syndrome.
Human molecular geneticsCorneal Cross-Linking for Brittle Cornea Syndrome.
CorneaBrittle Cornea Syndrome: Case Report with Novel Mutation in the PRDM5 Gene and Review of the Literature.
Case reports in ophthalmological medicineBrittle cornea syndrome: a case report and comparison with Ehlers Danlos syndrome.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusBrittle cornea syndrome ZNF469 mutation carrier phenotype and segregation analysis of rare ZNF469 variants in familial keratoconus.
Investigative ophthalmology & visual scienceAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome da córnea frágil.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome da córnea frágil
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- The Genetic and Epigenetic Architecture of Keratoconus: Emerging Pathways and Clinical Implications.
- Brittle cornea syndrome: Integrating unique presentations and novel management options.
- Brittle Cornea Syndrome Type 1 in Siblings: Severe Presentation in One and Misdiagnosis as Primary Congenital Glaucoma in the Other.
- Brittle Cornea Syndrome: Molecular Diagnosis and Management.
- [Ocular manifestations and genetic aspects of Ehlers-Danlos syndrome].
- Insight into genes responsible for cornea plana, megalocornea, keratoconus and brittle cornea syndrome.
- Clinical and Molecular Features of 11 Patients with Different Subtypes of Ehlers-Danlos Syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:90354(Orphanet)
- MONDO:0009242(MONDO)
- GARD:1019(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3508598(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
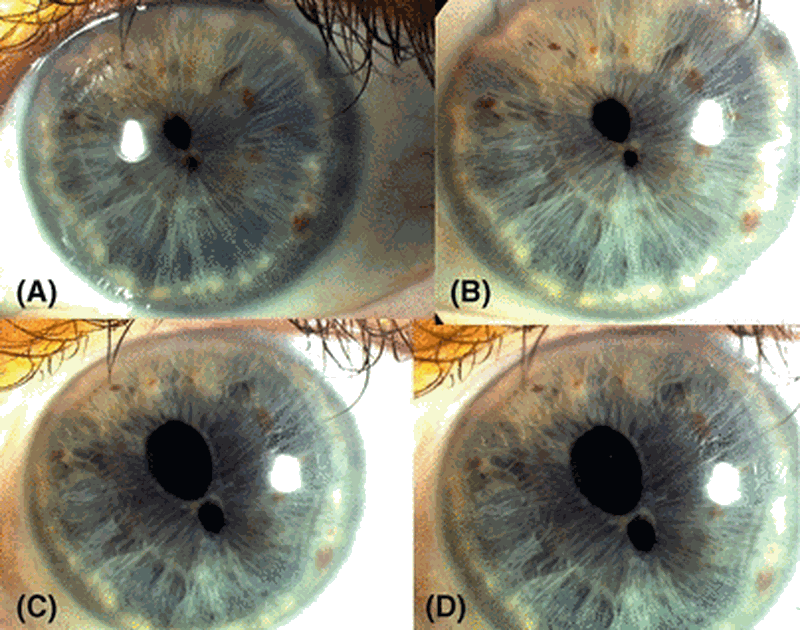
Síndrome da córnea frágil
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata