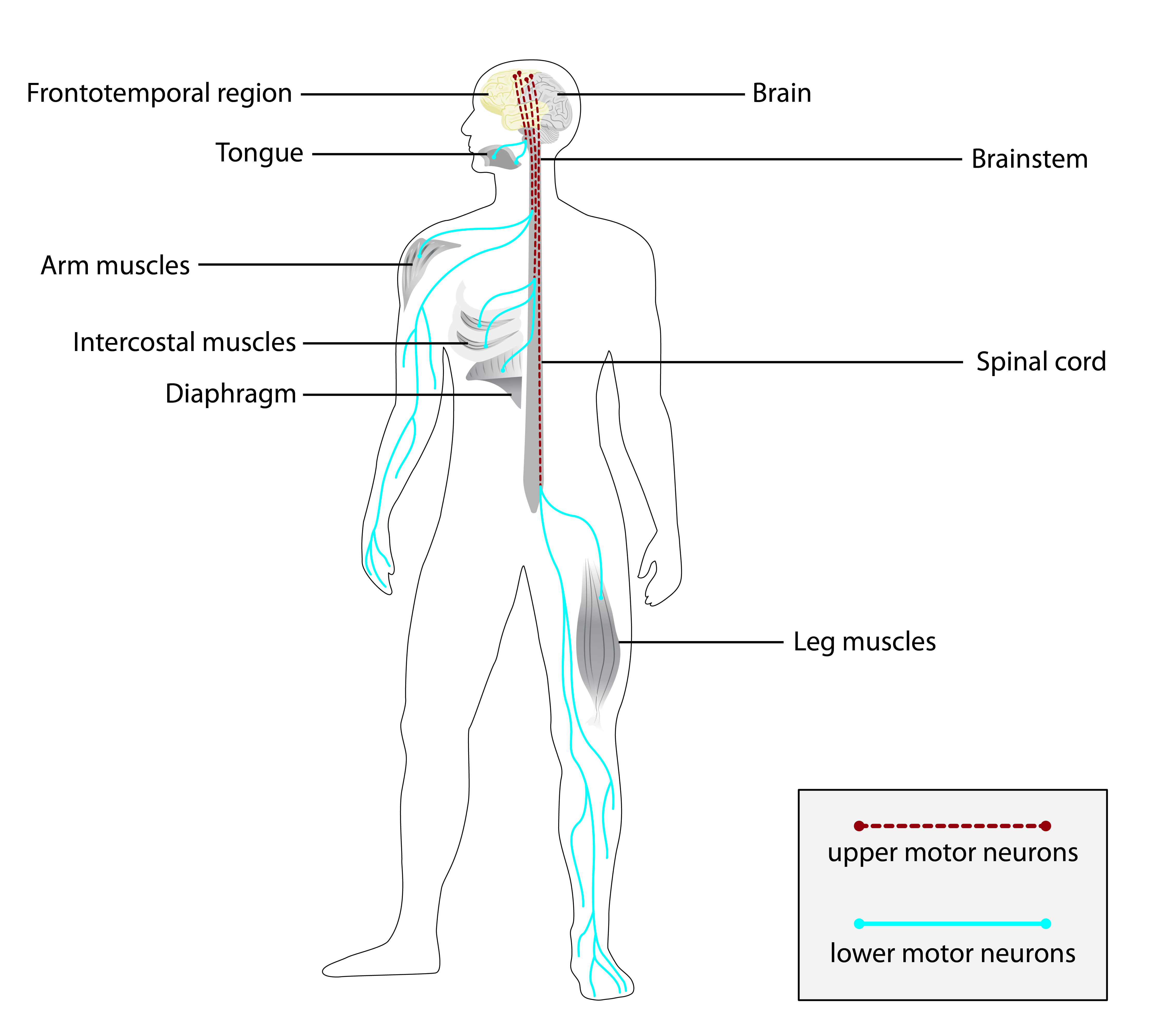
As paraplegias espásticas hereditárias (PHS) compreendem um grupo genética e clinicamente heterogêneo de doenças neurodegenerativas caracterizadas por espasticidade progressiva e hiperreflexia dos membros inferiores.
Introdução
O que você precisa saber de cara
As paraplegias espásticas hereditárias (PHS) compreendem um grupo genética e clinicamente heterogêneo de doenças neurodegenerativas caracterizadas por espasticidade progressiva e hiperreflexia dos membros inferiores.
Tem tratamento?
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 280 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 694 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
67 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, X-linked recessive.
May play a role as a negative regulatory factor in CD4-dependent T-cell activation
Cytoplasm, cytosolMembraneEndosome membraneGolgi apparatus, trans-Golgi network membrane
Spastic paraplegia 21, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG21 is associated with dementia and other central nervous system abnormalities. Subtle childhood abnormalities may be present, but the main features develop in early adulthood. The disease is slowly progressive, and cerebellar and extrapyramidal signs are also found in patients with advanced disease. Patients have a thin corpus callosum and white-matter abnormalities.
Involved in postimplantation and gastrulation stages of development. Involved in the nucleocytoplasmic shuttling of STAU2. Binds to DNA and RNA (By similarity)
NucleusCytoplasmCytoplasmic granuleChromosome
Component of the chaperonin-containing T-complex (TRiC), a molecular chaperone complex that assists the folding of actin, tubulin and other proteins upon ATP hydrolysis (PubMed:25467444, PubMed:36493755, PubMed:35449234, PubMed:37193829). The TRiC complex mediates the folding of WRAP53/TCAB1, thereby regulating telomere maintenance (PubMed:25467444). As part of the TRiC complex may play a role in the assembly of BBSome, a complex involved in ciliogenesis regulating transports vesicles to the cil
CytoplasmCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Neuropathy, hereditary sensory, with spastic paraplegia, autosomal recessive
A disease characterized by spastic paraplegia and progressive distal sensory neuropathy leading to mutilating ulcerations of the upper and lower limbs.
Acts as a component of the WASH core complex that functions as a nucleation-promoting factor (NPF) at the surface of endosomes, where it recruits and activates the Arp2/3 complex to induce actin polymerization, playing a key role in the fission of tubules that serve as transport intermediates during endosome sorting (PubMed:19922875, PubMed:20498093). May be involved in axonal outgrowth. Involved in cellular localization of ADRB2 (PubMed:23085491). Involved in cellular trafficking of BLOC-1 comp
Cytoplasm, cytosolEndoplasmic reticulumEarly endosome
Spastic paraplegia 8, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
One gap junction consists of a cluster of closely packed pairs of transmembrane channels, the connexons, through which materials of low MW diffuse from one cell to a neighboring cell. May play a role in myelination in central and peripheral nervous systems
Cell membraneCell junction, gap junction
Leukodystrophy, hypomyelinating, 2
An autosomal recessive hypomyelinating leukodystrophy with symptoms of Pelizaeus-Merzbacher disease. Clinically characterized by nystagmus, impaired motor development, ataxia, choreoathetotic movements, dysarthria, and progressive spasticity.
Regulatory subunit of the Rab3 GTPase-activating (Rab3GAP) complex composed of RAB3GAP1 and RAB3GAP2, which accelerates the otherwise slow GTP hydrolysis catalyzed by Rab proteins (PubMed:9733780, PubMed:39779760). The complex has GTPase-activating protein (GAP) activity towards various Rab3 subfamily members (RAB3A, RAB3B, RAB3C and RAB3D), RAB5A and RAB43, and has guanine nucleotide exchange factor (GEF) activity towards RAB18 (PubMed:9733780, PubMed:39779760, PubMed:24891604). The Rab3GAP com
CytoplasmEndoplasmic reticulum
Martsolf syndrome 1
An autosomal recessive disease characterized by congenital cataracts, intellectual disability, and hypogonadism.
As part of AP-5, a probable fifth adaptor protein complex it may be involved in endosomal transport. According to PubMed:20613862 it is a putative helicase required for efficient homologous recombination DNA double-strand break repair
CytoplasmNucleus
Spastic paraplegia 48, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
GPI inositol-deacylase that catalyzes the remove of the acyl chain linked to the 2-OH position of inositol ring from the GPI-anchored protein (GPI-AP) in the endoplasmic reticulum (PubMed:24784135, PubMed:38167496). Initiates the post-attachment remodeling phase of GPI-AP biogenesis and participates in endoplasmic reticulum (ER)-to-Golgi transport of GPI-anchored protein (PubMed:24784135, PubMed:38167496)
Endoplasmic reticulum membrane
Neurodevelopmental disorder with dysmorphic features, spasticity, and brain abnormalities
An autosomal recessive disorder characterized by severely delayed global development, with hypotonia, impaired intellectual development, and poor or absent speech. Most patients have spasticity with limb hypertonia and brisk tendon reflexes. Additional features include non-specific dysmorphic facial features, structural brain abnormalities, and cortical visual impairment.
AMP deaminase plays a critical role in energy metabolism. Catalyzes the deamination of AMP to IMP and plays an important role in the purine nucleotide cycle
Pontocerebellar hypoplasia 9
A form of pontocerebellar hypoplasia, a disorder characterized by structural defects of the pons and cerebellum, evident upon brain imaging. PCH9 features include severely delayed psychomotor development, progressive microcephaly, spasticity, seizures, and brain abnormalities, including brain atrophy, thin corpus callosum, and delayed myelination.
Plays a role in fibroblast growth factor-mediated signaling cascades that lead to the activation of MAP kinases. Promotes neurite outgrowth via FGFR1-mediated activation of downstream MAP kinases. Promotes an increase both in neurite number and in neurite length. May play a role in cell-cell adhesion and cell guidance via its interaction with ADGRL1/LPHN1 and ADGRL3
Cell membraneEndoplasmic reticulum membraneCytoplasmic vesicle membraneCytoplasm, perinuclear regionCell junction, focal adhesionSecretedCell projection, neuron projectionCell junction
Plays a role in the normal dynamic function of the endoplasmic reticulum (ER) and its associated microtubules (PubMed:23479643, PubMed:27813252). Required for secretory cargo traffic from the endoplasmic reticulum to the Golgi apparatus (PubMed:21478858)
Endoplasmic reticulum
Mitochondrial protein involved in the maturation of mitochondrial [4Fe-4S]-proteins in the late stage of the iron-sulfur cluster assembly pathway (PubMed:22323289, PubMed:23462291). Operates in cooperation with ISCA2 in the maturation of [4Fe-4S] proteins (PubMed:30269484) Involved in the maturation of mitochondrial 2Fe-2S proteins in the late stage of the iron-sulfur cluster assembly pathway
Mitochondrion
Multiple mitochondrial dysfunctions syndrome 3
A severe disorder of systemic energy metabolism, resulting in weakness, respiratory failure, lack of neurologic development, lactic acidosis, hyperglycinemia and early death. Some patients show failure to thrive, pulmonary hypertension, hypotonia and irritability. Biochemical features include severe combined deficiency of the 2-oxoacid dehydrogenases, defective lipoic acid synthesis and reduction in activity of mitochondrial respiratory chain complexes.
E3 ubiquitin-protein ligase involved in Golgi membrane fusion and regulation of small GTPases (PubMed:15254018, PubMed:21988917, PubMed:22036506, PubMed:37537642, PubMed:38332367). Acts as a regulator of Golgi membrane dynamics during the cell cycle: recruited to Golgi membrane by Rab proteins and regulates postmitotic Golgi membrane fusion (PubMed:21988917). Acts by mediating ubiquitination during mitotic Golgi disassembly, ubiquitination serving as a signal for Golgi reassembly later, after ce
Golgi apparatus, Golgi stack membraneCytoplasmEndoplasmic reticulum
ATP-dependent microtubule severing protein that specifically recognizes and cuts microtubules that are polyglutamylated (PubMed:11809724, PubMed:15716377, PubMed:16219033, PubMed:17389232, PubMed:20530212, PubMed:22637577, PubMed:26875866). Preferentially recognizes and acts on microtubules decorated with short polyglutamate tails: severing activity increases as the number of glutamates per tubulin rises from one to eight, but decreases beyond this glutamylation threshold (PubMed:26875866). Seve
MembraneEndoplasmic reticulumMidbodyCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeletonCytoplasm, perinuclear regionNucleusCytoplasm, cytoskeleton, spindleCytoplasmCell projection, axonEndoplasmic reticulum membraneNucleus membraneLipid dropletEndosome
Spastic paraplegia 4, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Kinesin is a microtubule-associated force-producing protein that plays a role in organelle transport. The light chain functions in coupling of cargo to the heavy chain or in the modulation of its ATPase activity (Probable). Through binding with PLEKHM2 and ARL8B, recruits kinesin-1 to lysosomes and hence direct lysosomes movement toward microtubule plus ends (PubMed:22172677)
Cytoplasm, cytoskeletonLysosome membrane
Spastic paraplegia, optic atrophy, and neuropathy
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPOAN is characterized by spastic paraplegia with progressive joint contractures and spine deformities, loss of independent ambulation by age 10 years, sub-normal vision secondary to congenital optic atrophy, and neuropathy. Inheritance is autosomal recessive.
Mitochondrial outer membrane GTPase that mediates mitochondrial clustering and fusion (PubMed:11181170, PubMed:11950885, PubMed:19889647, PubMed:26214738, PubMed:28114303). Mitochondria are highly dynamic organelles, and their morphology is determined by the equilibrium between mitochondrial fusion and fission events (PubMed:28114303). Overexpression induces the formation of mitochondrial networks (PubMed:28114303). Membrane clustering requires GTPase activity and may involve a major rearrangeme
Mitochondrion outer membrane
Charcot-Marie-Tooth disease, axonal, type 2A2B
An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2A2B is a severe form with autosomal recessive inheritance.
Necessary for the fragmentation of Golgi stacks during mitosis and for their reassembly after mitosis. Involved in the formation of the transitional endoplasmic reticulum (tER). The transfer of membranes from the endoplasmic reticulum to the Golgi apparatus occurs via 50-70 nm transition vesicles which derive from part-rough, part-smooth transitional elements of the endoplasmic reticulum (tER). Vesicle budding from the tER is an ATP-dependent process. The ternary complex containing UFD1, VCP and
Cytoplasm, cytosolEndoplasmic reticulumNucleusCytoplasm, Stress granule
Inclusion body myopathy with early-onset Paget disease with or without frontotemporal dementia 1
An autosomal dominant disease characterized by disabling muscle weakness clinically resembling to limb girdle muscular dystrophy, osteolytic bone lesions consistent with Paget disease, and premature frontotemporal dementia. Clinical features show incomplete penetrance.
Phosphatidylserine (PS) lipase that mediates the hydrolysis of phosphatidylserine to generate lysophosphatidylserine (LPS) (By similarity). LPS constitutes a class of signaling lipids that regulates immunological and neurological processes (By similarity). Has no activity towards diacylglycerol, triacylglycerol or lysophosphatidylserine lipase (PubMed:25290914). Also has monoacylglycerol lipase activity, with preference for 1-(9Z,12Z-octadecadienoyl)-glycerol (1-LG) and 2-glyceryl-15-deoxy-Delta
Membrane
Spastic paraplegia 86, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG86 is an autosomal recessive form associated with impaired intellectual development, poor or absent speech, and behavioral abnormalities. Brain imaging shows thin corpus callosum and white matter abnormalities. Rare patients may have seizures.
Component of the ESCRT-I complex, a regulator of vesicular trafficking process. Required for the sorting of endocytic ubiquitinated cargos into multivesicular bodies. May be involved in cell growth and differentiation
Late endosome membraneNucleus
Spastic paraplegia 53, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. Complicated forms are recognized by additional variable features including spastic quadriparesis, seizures, dementia, amyotrophy, extrapyramidal disturbance, cerebral or cerebellar atrophy, optic atrophy, and peripheral neuropathy, as well as by extra neurological manifestations. SPG53 is characterized by pronounced early onset spastic paraparesis of upper and lower limbs, mild intellectual disability, kyphosis, pectus carinatum and hypertrichosis.
Palmitoyl thioesterase specifically expressed in the endoplasmic reticulum of neurons. Modulates the trafficking of the glutamate receptor, AMPAR, to plasma membrane through depalmitoylation of GRIA1 (PubMed:30135643). Also regulates AMPR trafficking through the regulation of SACM1L phosphatidylinositol-3-phosphatase activity by interaction in a malonyl-CoA dependent manner (By similarity). Binds malonyl-CoA and couples malonyl-CoA to ceramide levels, necessary for proper spine maturation and co
Cell projection, dendriteCell projection, axonEndoplasmic reticulum membrane
Spastic paraplegia 73, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Part of a mitoribosome-associated quality control pathway that prevents aberrant translation by responding to interruptions during elongation (PubMed:33243891). As heterodimer with MTRES1, ejects the unfinished nascent chain and peptidyl transfer RNA (tRNA), respectively, from stalled ribosomes. Recruitment of mitoribosome biogenesis factors to these quality control intermediates suggests additional roles for MTRES1 and MTRF during mitoribosome rescue (PubMed:33243891)
Mitochondrion
Combined oxidative phosphorylation deficiency 7
A mitochondrial disease resulting in encephalomyopathy. Clinical manifestations include psychomotor delay and regression, ataxia, optic atrophy, nystagmus and muscle atrophy and weakness.
Component of the adaptor protein complex 4 (AP-4). Adaptor protein complexes are vesicle coat components involved both in vesicle formation and cargo selection. They control the vesicular transport of proteins in different trafficking pathways (PubMed:10066790, PubMed:10436028). AP-4 forms a non clathrin-associated coat on vesicles departing the trans-Golgi network (TGN) and may be involved in the targeting of proteins from the trans-Golgi network (TGN) to the endosomal-lysosomal system. It is a
Golgi apparatus, trans-Golgi network membrane
Spastic paraplegia 52, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. SPG52 is characterized by neonatal hypotonia that progresses to hypertonia and spasticity, and severe intellectual disability with poor or absent speech development. Some patients may have seizures.
E3 ubiquitin-protein ligase that plays an essential role in stimulus-induced inositol 1,4,5-trisphosphate receptor type 1 (ITPR1) ubiquitination and degradation via the endoplasmic reticulum-associated degradation (ERAD) pathway. Also involved in ITPR1 turnover in resting cells. Selectively inhibits the TLR3-triggered innate immune response by promoting the 'Lys-48'-linked polyubiquitination and degradation of TLR3 (PubMed:31076723)
Endoplasmic reticulum membrane
Ataxia, sensory, 1, autosomal dominant
A rare disease characterized by progressive ataxia caused by degeneration of the posterior columns of the spinal cord. Affected individuals have a reduced ability to feel pain, temperature and vibration, particularly in the hands and feet. Their most prominent feature is an ataxic gait resulting from a severe loss of proprioception. Thus, patients rely on visual cues for maintaining proper body posture, such that they are unable to remain upright if their eyes are closed (Romberg sign).
Adhesion molecule that mediates interactions between myelinating cells and neurons by binding to neuronal sialic acid-containing gangliosides and to the glycoproteins RTN4R and RTN4RL2 (By similarity). Not required for initial myelination, but seems to play a role in the maintenance of normal axon myelination. Protects motoneurons against apoptosis, also after injury; protection against apoptosis is probably mediated via interaction with neuronal RTN4R and RTN4RL2. Required to prevent degenerati
Cell membraneMembrane raft
Spastic paraplegia 75, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG75 is characterized by onset in early childhood and is associated with mild to moderate cognitive impairment.
Catalyzes the hydrolysis of nucleoside triphosphates (NTPs) and diphosphates (NDPs) (Probable) (PubMed:8529670, PubMed:8626624, PubMed:8955160, PubMed:8996251). The enzyme sequentially removes phosphate groups in two successive steps, converting NTPs to nucleoside monophosphates (NMPs) via NDP intermediates (Probable) (PubMed:8529670, PubMed:8626624, PubMed:8955160, PubMed:8996251). This activity contributes to the regulation of extracellular levels of nucleotides (Probable) (PubMed:8529670, Pub
MembraneMembrane, caveola
Spastic paraplegia 64, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
A cytochrome P450 monooxygenase involved in the metabolism of endogenous oxysterols and steroid hormones, including neurosteroids (PubMed:10588945, PubMed:24491228). Mechanistically, uses molecular oxygen inserting one oxygen atom into a substrate, and reducing the second into a water molecule, with two electrons provided by NADPH via cytochrome P450 reductase (CPR; NADPH-ferrihemoprotein reductase) (PubMed:10588945, PubMed:24491228). Catalyzes the hydroxylation of carbon hydrogen bonds of stero
Endoplasmic reticulum membraneMicrosome membrane
Spastic paraplegia 5A, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
A cytochrome P450 monooxygenase involved in the metabolism of arachidonic acid and its conjugates (PubMed:14660610, PubMed:24563460). Mechanistically, uses molecular oxygen inserting one oxygen atom into a substrate, and reducing the second into a water molecule, with two electrons provided by NADPH via cytochrome P450 reductase (CPR; NADPH-ferrihemoprotein reductase) (PubMed:14660610, PubMed:24563460). Acts as an omega and omega-1 hydroxylase for arachidonic acid and possibly for other long cha
Endoplasmic reticulum membraneMicrosome membraneMitochondrion inner membrane
Spastic paraplegia 56, autosomal recessive, with or without pseudoxanthoma elasticum
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. Complicated forms are recognized by additional variable features including spastic quadriparesis, seizures, dementia, amyotrophy, extrapyramidal disturbance, cerebral or cerebellar atrophy, optic atrophy, and peripheral neuropathy, as well as by extra neurological manifestations. In SPG56, upper limbs are often also affected. Some SPG56 patients may have a subclinical axonal neuropathy; others also have pseudoxanthoma elasticum.
Acetyl-CoA transporter that mediates active acetyl-CoA import through the endoplasmic reticulum (ER) membrane into the ER lumen where specific ER-based acetyl-CoA:lysine acetyltransferases are responsible for the acetylation of ER-based protein substrates, such as BACE1 (PubMed:20826464, PubMed:24828632). Necessary for O-acetylation of gangliosides (PubMed:9096318)
Endoplasmic reticulum membrane
Spastic paraplegia 42, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Component of the ESCRT-I complex, a regulator of vesicular trafficking process (PubMed:21757351, PubMed:22405001, PubMed:31203368). Binds to ubiquitinated cargo proteins and is required for the sorting of endocytic ubiquitinated cargos into multivesicular bodies (MVBs) (PubMed:21757351, PubMed:22405001). Plays a role in the proteasomal degradation of ubiquitinated cell-surface proteins, such as EGFR and BST2 (PubMed:22405001, PubMed:24284069, PubMed:31203368)
Cytoplasm, cytosolEndosome
Spastic paraplegia 80, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Component of the ERLIN1/ERLIN2 complex which mediates the endoplasmic reticulum-associated degradation (ERAD) of inositol 1,4,5-trisphosphate receptors (IP3Rs). Involved in regulation of cellular cholesterol homeostasis by regulation the SREBP signaling pathway (PubMed:37683630). Binds cholesterol and may promote ER retention of the SCAP-SREBF complex (PubMed:24217618) (Microbial infection) Required early in hepatitis C virus (HCV) infection to initiate RNA replication, and later in the infectio
Endoplasmic reticulum membrane
Spastic paraplegia 62, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Non-lysosomal glucosylceramidase that catalyzes the hydrolysis of glucosylceramides/GlcCers (such as beta-D-glucosyl-(1<->1')-N-acylsphing-4-enine) to free glucose and ceramides (such as N-acylsphing-4-enine) (PubMed:17105727, PubMed:30308956, PubMed:32144204). GlcCers are membrane glycosphingolipids that have a wide intracellular distribution (By similarity). They are the main precursors of more complex glycosphingolipids that play a role in cellular growth, differentiation, adhesion, signaling
Endoplasmic reticulum membraneGolgi apparatus membrane
Spastic paraplegia 46, autosomal recessive
A neurodegenerative disorder characterized by onset in childhood of slowly progressive spastic paraplegia and cerebellar signs. Some patients have cognitive impairment, cataracts, and cerebral, cerebellar, and corpus callosum atrophy on brain imaging.
Broad specificity cytosolic 5'-nucleotidase that catalyzes the dephosphorylation of 6-hydroxypurine nucleoside 5'-monophosphates (PubMed:10092873, PubMed:12907246, PubMed:1659319, PubMed:9371705). In addition, possesses a phosphotransferase activity by which it can transfer a phosphate from a donor nucleoside monophosphate to an acceptor nucleoside, preferably inosine, deoxyinosine and guanosine (PubMed:1659319, PubMed:9371705). Has the highest activities for IMP and GMP followed by dIMP, dGMP a
Cytoplasm, cytosol
Spastic paraplegia 45, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. Some SPG45 patients manifest intellectual disability, contractures and learning disability.
Acts as a positive regulator of ERK phosphorylation downstream of fibroblast growth factor-receptor activation (PubMed:23862974, PubMed:28157540). Involved in the regulation of both caspase-dependent apoptosis and caspase-independent cell death (PubMed:15178406). In the skin, it plays a predominant role in suppressing caspase-dependent apoptosis in response to UV stress in a range of dermal cell types (PubMed:28157540)
CytoplasmCell membraneApical cell membraneBasolateral cell membraneCell junction
Congenital anomalies of the kidney and urinary tract 1
A disorder encompassing a broad spectrum of renal and urinary tract malformations that include renal agenesis, kidney hypodysplasia, multicystic kidney dysplasia, duplex collecting system, posterior urethral valves and ureter abnormalities. Congenital anomalies of kidney and urinary tract are the commonest cause of chronic kidney disease in children.
Component of the ERLIN1/ERLIN2 complex which mediates the endoplasmic reticulum-associated degradation (ERAD) of inositol 1,4,5-trisphosphate receptors (IP3Rs) such as ITPR1 (PubMed:17502376, PubMed:19240031). Promotes sterol-accelerated ERAD of HMGCR probably implicating an AMFR/gp78-containing ubiquitin ligase complex (PubMed:21343306). Involved in regulation of cellular cholesterol homeostasis by regulation the SREBP signaling pathway. May promote ER retention of the SCAP-SREBF complex (PubMe
Endoplasmic reticulum membrane
Spastic paraplegia 18B, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG18B is a severe form with onset in early childhood. Most affected individuals have severe psychomotor retardation. Some may develop significant joint contractures.
Catalyzes the reversible transamination between alanine and 2-oxoglutarate to form pyruvate and glutamate
Neurodevelopmental disorder with spastic paraplegia and microcephaly
An autosomal recessive syndrome characterized by severe psychomotor developmental delay, dysarthria, walking difficulties, moderately to severely impaired intellectual development, poor or absent speech, and progressive microcephaly.
Iron-dependent dioxygenase that catalyzes the conversion of 4-hydroxyphenylpyruvate (4-HPPA) to 4-hydroxymandelate (4-HMA) in the mitochondria, one of the steps in the biosynthesis of coenzyme Q10 from tyrosine
Mitochondrion
Neurodevelopmental disorder with progressive spasticity and brain white matter abnormalities
An autosomal recessive neurodevelopmental disorder characterized by developmental delay manifesting in infancy, inability to walk independently, mild to severe intellectual disability, poor overall growth, progressive microcephaly, and axial hypotonia. Additional variable features include brainstem and cerebellar involvement, seizures, joint contractures, ocular disturbances, episodic respiratory failure, and facial dysmorphism.
Catalytic component of the m-AAA protease, a protease that plays a key role in proteostasis of inner mitochondrial membrane proteins, and which is essential for axonal and neuron development (PubMed:11549317, PubMed:28396416, PubMed:31097542, PubMed:9635427). SPG7 possesses both ATPase and protease activities: the ATPase activity is required to unfold substrates, threading them into the internal proteolytic cavity for hydrolysis into small peptide fragments (By similarity). The m-AAA protease ex
Mitochondrion inner membrane
Spastic paraplegia 7, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG7 is a complex form. Additional clinical features are cerebellar syndrome, supranuclear palsy, and cognitive impairment, particularly disturbance of attention and executive functions.
Positively regulates SLC1A1/EAAC1-mediated glutamate transport by increasing its affinity for glutamate in a PKC activity-dependent manner. Promotes the catalytic efficiency of SLC1A1/EAAC1 probably by reducing its interaction with ARL6IP5, a negative regulator of SLC1A1/EAAC1-mediated glutamate transport (By similarity). Plays a role in the formation and stabilization of endoplasmic reticulum tubules (PubMed:24262037). Negatively regulates apoptosis, possibly by modulating the activity of caspa
Endomembrane systemEndoplasmic reticulum membraneEndoplasmic reticulum
Spastic paraplegia 61, autosomal recessive
A complicated form of spastic paraplegia with polysensory and motor neuropathy. Spastic paraplegia is a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Phospholipase A1 (PLA1) that hydrolyzes ester bonds at the sn-1 position of glycerophospholipids producing a free fatty acid and a lysophospholipid (Probable) (PubMed:20359546, PubMed:22922100). Prefers phosphatidate (1,2-diacyl-sn-glycero-3-phosphate, PA) as substrate in vitro, but can efficiently hydrolyze phosphatidylinositol (1,2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol), PI), as well as a range of other glycerophospholipid substrates such as phosphatidylcholine (1,2-diacyl-sn-glycero-3-
Cytoplasm
Spastic paraplegia 28, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. Some SPG28 patients also have distal sensory impairment.
Chaperonin implicated in mitochondrial protein import and macromolecular assembly. Together with Hsp10, facilitates the correct folding of imported proteins. May also prevent misfolding and promote the refolding and proper assembly of unfolded polypeptides generated under stress conditions in the mitochondrial matrix (PubMed:11422376, PubMed:1346131). The functional units of these chaperonins consist of heptameric rings of the large subunit Hsp60, which function as a back-to-back double ring. In
Mitochondrion matrix
Spastic paraplegia 13, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Inhibits amyloid precursor protein processing, probably by blocking BACE1 activity (PubMed:15286784). Enhances trafficking of the glutamate transporter SLC1A1/EAAC1 from the endoplasmic reticulum to the cell surface (By similarity). Plays a role in the translocation of SLC2A4/GLUT4 from intracellular membranes to the cell membrane which facilitates the uptake of glucose into the cell (By similarity)
Endoplasmic reticulum membraneSarcoplasmic reticulum membraneCell membraneCell membrane, sarcolemmaCell membrane, sarcolemma, T-tubuleCytoplasm, myofibril, sarcomere, Z lineCytoplasm, cytoskeleton
Spastic paraplegia 12, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Microtubule-dependent motor required for slow axonal transport of neurofilament proteins (NFH, NFM and NFL). Can induce formation of neurite-like membrane protrusions in non-neuronal cells in a ZFYVE27-dependent manner. The ZFYVE27-KIF5A complex contributes to the vesicular transport of VAPA, VAPB, SURF4, RAB11A, RAB11B and RTN3 proteins in neurons. Required for anterograde axonal transportation of MAPK8IP3/JIP3 which is essential for MAPK8IP3/JIP3 function in axon elongation
Cytoplasm, perinuclear regionCytoplasm, cytoskeletonPerikaryon
Spastic paraplegia 10, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Neural cell adhesion molecule involved in the dynamics of cell adhesion and in the generation of transmembrane signals at tyrosine kinase receptors. During brain development, critical in multiple processes, including neuronal migration, axonal growth and fasciculation, and synaptogenesis. In the mature brain, plays a role in the dynamics of neuronal structure and function, including synaptic plasticity
Cell membraneCell projection, growth coneCell projection, axonCell projection, dendrite
Hydrocephalus, congenital, X-linked
An X-linked recessive form of congenital hydrocephalus, a disease characterized by in utero onset of enlarged ventricles due to accumulation of ventricular cerebrospinal fluid. HYCX is the most common inherited form and occurs in approximately 1/30000 male births. The primary diagnostic criteria of intellectual disability and enlarged cerebral ventricles are often accompanied by spastic paraparesis and adducted thumbs and, occasionally, visual defects or seizures. The most severe cases die pre- or perinatally with gross hydrocephalus and enlarged head circumference. Stenosis of the aqueduct of Sylvius is frequently associated with the disorder.
Promotes a prolonged MAP-kinase signaling by neurotrophins through activation of a Rap1-dependent mechanism. Provides a docking site for the CRKL-C3G complex, resulting in Rap1-dependent sustained ERK activation. May play an important role in regulating postsynaptic signal transduction through the syntrophin-mediated localization of receptor tyrosine kinases such as EPHA4. In cooperation with SNTA1 can enhance EPHA4-induced JAK/STAT activation. Plays a role in nerve growth factor (NGF)-induced r
MembraneLate endosome
Spastic paraplegia, intellectual disability, nystagmus, and obesity
An autosomal dominant syndrome characterized by rapid growth in infancy, obesity, global developmental delay, intellectual disability, spastic paraplegia, ocular defects, and dysmorphic facial features.
Acts on phosphatidylinositol (PtdIns) in the first committed step in the production of the second messenger inositol-1,4,5,-trisphosphate
CytoplasmCell membrane
Neurodevelopmental disorder with spasticity, hypomyelinating leukodystrophy, and brain abnormalities
A severe autosomal recessive disorder characterized by global developmental delay with impaired intellectual development and poor or absent speech, axial hypotonia, and peripheral spasticity and hyperreflexia. Brain imaging shows hypomyelination with decreased white matter volume, cerebral and cerebellar atrophy, and thin corpus callosum. Polymicrogyria may be observed in rare cases. Some patients have a primary immunodeficiency or gastrointestinal disturbances similar to inflammatory bowel disease.
Is responsible for the charging of tRNA(Phe) with phenylalanine in mitochondrial translation. To a lesser extent, also catalyzes direct attachment of m-Tyr (an oxidized version of Phe) to tRNA(Phe), thereby opening the way for delivery of the misacylated tRNA to the ribosome and incorporation of ROS-damaged amino acid into proteins
Mitochondrion matrixMitochondrion
Combined oxidative phosphorylation deficiency 14
A severe multisystemic autosomal recessive disorder characterized by neonatal onset of global developmental delay, refractory seizures, and lactic acidosis. Biochemical studies show deficiencies of multiple mitochondrial respiratory enzymes.
Calcium-regulated non-lysosomal thiol-protease which catalyzes limited proteolysis of substrates involved in cytoskeletal remodeling and signal transduction (PubMed:19617626, PubMed:21531719, PubMed:2400579). Proteolytically cleaves CTBP1 at 'Asn-375', 'Gly-387' and 'His-409' (PubMed:23707407). Cleaves and activates caspase-7 (CASP7) (PubMed:19617626)
CytoplasmCell membrane
Spastic paraplegia 76, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Diacylglycerol (DAG) and triacylglycerol (TAG) lipase required for proper lipid homeostasis in the central nervous system (PubMed:29278326, PubMed:37832604). It cooperates with PNPLA2/ATGL in neuronal TAG catabolism and hydrolyzes sn-1,3 DAG downstream of PNPLA2/ATGL (By similarity). In vitro, it also acts as a phospholipase that hydrolyzes preferentially phosphatidic acids, including 1,2-dioleoyl-sn-phosphatidic acid, phosphatidylcholine and phosphatidylethanolamine. Specifically binds to phosp
Cytoplasm, cytosolEndoplasmic reticulum-Golgi intermediate compartmentGolgi apparatus, cis-Golgi network
Spastic paraplegia 54, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. Complicated forms are recognized by additional variable features including spastic quadriparesis, seizures, dementia, amyotrophy, extrapyramidal disturbance, cerebral or cerebellar atrophy, optic atrophy, and peripheral neuropathy, as well as by extra neurological manifestations. SPG54 patients have delayed psychomotor development, intellectual disability, and early-onset spasticity of the lower limbs. Brain MRI shows a thin corpus callosum and periventricular white matter lesions, and an abnormal lipid peak due to accumulation of neutral lipids in certain brain regions.
Phosphatidylinositol 3-phosphate-binding protein required for the abscission step in cytokinesis: recruited to the midbody during cytokinesis and acts as a regulator of abscission. May also be required for efficient homologous recombination DNA double-strand break repair
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeMidbody
Spastic paraplegia 15, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG15 is a complex form associated with additional neurological symptoms such as cognitive deterioration or intellectual disability, axonal neuropathy, mild cerebellar signs, and, less frequently, a central hearing deficit, decreased visual acuity, or retinal degeneration.
Plays a crucial role in the formation of lipid droplets (LDs) which are storage organelles at the center of lipid and energy homeostasis (PubMed:19278620, PubMed:21533227, PubMed:30293840, PubMed:31708432). In association with LDAF1, defines the sites of LD formation in the ER (PubMed:31708432). Also required for growth and maturation of small nascent LDs into larger mature LDs (PubMed:27564575). Mediates the formation and/or stabilization of endoplasmic reticulum-lipid droplets (ER-LD) contacts
Endoplasmic reticulum membraneLipid droplet
Lipodystrophy, congenital generalized, 2
A form of congenital generalized lipodystrophy, a metabolic disorder characterized by a near complete absence of adipose tissue, extreme insulin resistance, hypertriglyceridemia, hepatic steatosis and early onset of diabetes. Inheritance is autosomal recessive.
Lipophagy receptor that plays an important role in lipid droplet (LD) turnover in motor neurons (PubMed:37443287). Localizes to LDs and interacts with components of the autophagy machinery, such as MAP1LC3A/C proteins to deliver LDs to autophagosomes for degradation via lipophagy (PubMed:37443287). Lipid transfer protein required for lipid droplet degradation, including by lipophagy (PubMed:38190532). Can bind and transfer all lipid species found in lipid droplets, from phospholipids to triglyce
CytoplasmMidbodyLipid droplet
Spastic paraplegia 20, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG20 is characterized by dysarthria, distal amyotrophy, mild developmental delay and short stature.
Component of the serine palmitoyltransferase multisubunit enzyme (SPT) that catalyzes the initial and rate-limiting step in sphingolipid biosynthesis by condensing L-serine and activated acyl-CoA (most commonly palmitoyl-CoA) to form long-chain bases (PubMed:19416851). The SPT complex is composed of SPTLC1, SPTLC2 or SPTLC3 and SPTSSA or SPTSSB. Within this complex, the heterodimer consisting of SPTLC1 and SPTLC2/SPTLC3 forms the catalytic core (PubMed:19416851). Within the SPT complex, SPTSSA s
Endoplasmic reticulum membrane
Spastic paraplegia 90A, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or weakness and stiffness may spread to other parts of the body. SPG90A affected individuals have motor impairment and progressive lower extremity spasticity as well as neurologic findings, cognitive impairment, and hearing loss.
Phospholipase B that deacylates intracellular phosphatidylcholine (PtdCho), generating glycerophosphocholine (GroPtdCho). This deacylation occurs at both sn-2 and sn-1 positions of PtdCho. Catalyzes the hydrolysis of several naturally occurring membrane-associated lipids (PubMed:11927584). Hydrolyzes lysophospholipids and monoacylglycerols, preferring the 1-acyl to the 2-acyl isomer. Does not catalyze hydrolysis of di- or triacylglycerols or fatty acid amides (PubMed:11927584)
Endoplasmic reticulum membrane
Spastic paraplegia 39, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG39 is associated with a motor axonopathy affecting upper and lower limbs and resulting in progressive wasting of distal upper and lower extremity muscles.
Involved in the biosynthesis of gangliosides GM2, GD2, GT2 and GA2 from GM3, GD3, GT3 and GA3, respectively
Golgi apparatus membrane
Spastic paraplegia 26, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG26 is a complicated form characterized by onset in the first 2 decades of life of gait abnormalities due to lower limb spasticity and muscle weakness. Some patients have upper limb involvement. Additional features include intellectual disability, peripheral neuropathy, dysarthria, cerebellar signs, extrapyramidal signs, and cortical atrophy. The disorder is slowly progressive.
Bifunctional enzyme that converts glutamate to glutamate 5-semialdehyde, an intermediate in the biosynthesis of proline, ornithine and arginine
MitochondrionMitochondrion matrix
Cutis laxa, autosomal recessive, 3A
A syndrome characterized by facial dysmorphism with a progeroid appearance, large and late-closing fontanel, cutis laxa, joint hyperlaxity, athetoid movements and hyperreflexia, pre- and postnatal growth retardation, intellectual deficit, developmental delay, and ophthalmologic abnormalities.
Ethanolamine-phosphate cytidylyltransferase that catalyzes the second step in the synthesis of phosphatidylethanolamine (PE) from ethanolamine via the CDP-ethanolamine pathway (PubMed:31637422, PubMed:9083101). Phosphatidylethanolamine is a dominant inner-leaflet phospholipid in cell membranes, where it plays a role in membrane function by structurally stabilizing membrane-anchored proteins, and participates in important cellular processes such as cell division, cell fusion, blood coagulation, a
Spastic paraplegia 82, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG82 is a complicated form characterized by global developmental delay with regression, spastic para- or tetraparesis, epilepsy and progressive cerebral and cerebellar atrophy.
Deubiquitinase that plays a role in the regulation of several processes such as maintenance of synaptic function, cardiac function, inflammatory response or osteoclastogenesis (PubMed:22212137, PubMed:23359680). Abrogates the ubiquitination of multiple proteins including WWTR1/TAZ, EGFR, HIF1A and beta-site amyloid precursor protein cleaving enzyme 1/BACE1 (PubMed:22212137, PubMed:25615526). In addition, recognizes and hydrolyzes a peptide bond at the C-terminal glycine of ubiquitin to maintain
CytoplasmEndoplasmic reticulum membrane
Parkinson disease 5
A complex neurodegenerative disorder with manifestations ranging from typical Parkinson disease to dementia with Lewy bodies. Clinical features include parkinsonian symptoms (resting tremor, rigidity, postural instability and bradykinesia), dementia, diffuse Lewy body pathology, autonomic dysfunction, hallucinations and paranoia.
Component of the adaptor protein complex 4 (AP-4). Adaptor protein complexes are vesicle coat components involved both in vesicle formation and cargo selection. They control the vesicular transport of proteins in different trafficking pathways (PubMed:10066790, PubMed:10436028). AP-4 forms a non clathrin-associated coat on vesicles departing the trans-Golgi network (TGN) and may be involved in the targeting of proteins from the trans-Golgi network (TGN) to the endosomal-lysosomal system. It is a
Golgi apparatus, trans-Golgi network membrane
Spastic paraplegia 51, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. SPG51 is a non-progressive disorder of movement and/or posture resulting from defects in the developing central nervous system. Affected individuals manifest motor and posture impairments often associated with epilepsy and disturbances of cognition, behavior, sensation, and communication.
Component of the adaptor protein complex 4 (AP-4). Adaptor protein complexes are vesicle coat components involved both in vesicle formation and cargo selection. They control the vesicular transport of proteins in different trafficking pathways (PubMed:10066790, PubMed:10436028, PubMed:11139587, PubMed:11802162, PubMed:20230749). AP-4 forms a non clathrin-associated coat on vesicles departing the trans-Golgi network (TGN) and may be involved in the targeting of proteins from the trans-Golgi netwo
Golgi apparatus, trans-Golgi network membraneEarly endosome
Spastic paraplegia 50, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG50 affected individuals present postnatally with early infantile hypotonia, delayed psychomotor development, strabismus, lack of independent walking and severe intellectual disability. They develop progressive spasticity of all limbs with generalized hypertonia, hyperreflexia, and extensor plantar responses by the end of the first year of life. Speech is absent or limited. Pseudobulbar signs, such as drooling, stereotypic laughter, and exaggerated jaw jerk, are part of the clinical picture.
Atlastin-1 (ATL1) is a membrane-anchored GTPase that mediates the GTP-dependent fusion of endoplasmic reticulum (ER) membranes, maintaining the continuous ER network. It facilitates the formation of three-way junctions where ER tubules intersect (PubMed:14506257, PubMed:18270207, PubMed:19665976, PubMed:27619977, PubMed:34817557, PubMed:38509071). Two atlastin-1 on neighboring ER tubules bind GTP and form loose homodimers through the GB1/RHD3-type G domains and 3HB regions. Upon GTP hydrolysis,
Endoplasmic reticulum membraneGolgi apparatus membraneCell projection, axon
Spastic paraplegia 3, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Catalyzes the hydroxylation of free fatty acids at the C-2 position to produce 2-hydroxy fatty acids, which are building blocks of sphingolipids and glycosphingolipids common in neural tissue and epidermis (PubMed:15337768, PubMed:15863841, PubMed:17355976, PubMed:22517924). FA2H is stereospecific for the production of (R)-2-hydroxy fatty acids (PubMed:22517924). Plays an essential role in the synthesis of galactosphingolipids of the myelin sheath (By similarity). Responsible for the synthesis o
Endoplasmic reticulum membraneMicrosome membrane
Spastic paraplegia 35, autosomal recessive, with or without neurodegeneration
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG35 is a complicated form characterized by childhood onset of gait difficulties. It has a rapid progression and many patients become wheelchair-bound as young adults. Patients manifest cognitive decline associated with leukodystrophy. Other variable neurologic features, such as dystonia, optic atrophy, and seizures may also occur.
Required for endoplasmic reticulum (ER) network formation, shaping and remodeling; it links ER tubules to the cytoskeleton. May also enhance the cell surface expression of odorant receptors (PubMed:20200447). May play a role in long-term axonal maintenance (PubMed:24478229)
MembraneMitochondrion membraneEndoplasmic reticulum
Spastic paraplegia 31, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Probably plays a role as positive regulator of autophagy
Neuropathy, hereditary sensory and autonomic, 9, with developmental delay
A form of hereditary sensory and autonomic neuropathy, a genetically and clinically heterogeneous group of disorders characterized by degeneration of dorsal root and autonomic ganglion cells, and by sensory and/or autonomic abnormalities. HSAN9 is characterized by global developmental delay and intellectual disability, axial and appendicular hypotonia, dysarthria, and an abnormal gait that is often described as ataxic. Other features may include peripheral neuropathy, hyporeflexia, and autonomic dysfunction. Affected individuals also have dysmorphic features, thin corpus callosum on brain imaging, and episodes of central apnea, which may be fatal.
Ethanolaminephosphotransferase that catalyzes the transfer of phosphoethanolamine (PE) from CDP-ethanolamine to lipid acceptors, the final step in the synthesis of PE via the 'Kennedy' pathway (PubMed:17132865, PubMed:28052917, PubMed:29500230). PE is the second most abundant phospholipid of membranes in mammals and is involved in various membrane-related cellular processes (PubMed:17132865). The enzyme is critical for the synthesis of several PE species and also catalyzes the synthesis of plasm
Endoplasmic reticulum membrane
Spastic paraplegia 81, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG81 is a complicated form characterized by white matter abnormalities, hypomyelination with progressive white matter loss, delayed motor development, progressive spasticity, and impaired intellectual development and speech delay. Additional features may include bifid uvula, microcephaly, seizures, and variable ocular anomalies.
Acts as a Mg(2+) transporter. Can also transport other divalent cations such as Fe(2+), Sr(2+), Ba(2+), Zn(2+) and Co(2+) but to a much less extent than Mg(2+) (By similarity)
Cell membraneEarly endosome
Spastic paraplegia 6, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body.
Kinesin motor with a plus-end-directed microtubule motor activity (By similarity). It is required for anterograde axonal transport of synaptic vesicle precursors (PubMed:33880452). Also required for neuronal dense core vesicles (DCVs) transport to the dendritic spines and axons. The interaction calcium-dependent with CALM1 increases vesicle motility and interaction with the scaffolding proteins PPFIA2 and TANC2 recruits DCVs to synaptic sites
Cytoplasm, cytoskeletonCell projection, neuron projectionCell projection, axonCytoplasm, perinuclear regionSynapseCytoplasmic vesicle, secretory vesicle, neuronal dense core vesicle membrane
Spastic paraplegia 30A, autosomal dominant
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. Some SPG30A patients have a pure form of the disorder, limited to spastic paraplegia, whereas others may have a complicated form that includes additional features such as cognitive dysfunction, learning disabilities, peripheral sensorimotor neuropathy, urinary sphincter problems, and/or cerebellar atrophy. SPG30A is characterized by onset in the first or second decades of unsteady spastic gait and hyperreflexia of the lower limbs. Inheritance is autosomal dominant.
Motor required for the retrograde transport of Golgi vesicles to the endoplasmic reticulum. Has a microtubule plus end-directed motility
Cytoplasm, cytoskeleton
Spastic ataxia 2, autosomal recessive
A neurologic disorder characterized by cerebellar ataxia, dysarthria, and variable spasticity of the lower limbs. Cognition is not affected.
Medicamentos e terapias
Mecanismo: Ceramide glucosyltransferase inhibitor
Variantes genéticas (ClinVar)
168 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 20,188 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
106 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Paraplegia espástica hereditária
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
20 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Mostrando amostra de 200 publicações de um total de 1.488
Inhibition of FicD-mediated AMPylation and deAMPylation by isoprenoid diphosphates.
FicD regulates Unfolded Protein Response (UPR) through reversible AMPylation and deAMPylation of BiP, an HSP70 chaperone and master regulator of the UPR. FicD activity is regulated by endoplasmic reticulum-stress, catalyzing BiP AMPylation under low stress conditions to hold inactive chaperone in reserve. In stressed cells, FicD deAMPylates BiP, acutely increasing its active pool to assist in protein folding. Variants in UPR machinery, including those in the FicD gene, are linked to hereditary diseases. Despite the known role of FicD in UPR, in-vivo regulation of its activity remains elusive, and identifying metabolites that alter FicD activity could prove useful pharmaceutically. We applied an unbiased high-throughput screening platform, known as Mass spectrometry Integrated with equilibrium Dialysis for the discovery of Allostery Systematically (MIDAS), to identify small molecule metabolites that might regulate FicD activity. MIDAS revealed interactions between FicD and two mevalonate pathway intermediates: geranyl-pyrophosphate and farnesyl-pyrophosphate. Biochemical characterization indicates that both potently inhibit FicD-mediated AMPylation and deAMPylation. The crystal structure of FicD bound to farnesyl-pyrophosphate demonstrates a competitive inhibition mechanism, with the pyrophosphate adopting the alpha and beta phosphate positions of adenosine triphosphate (ATP) and the hydrocarbon chain filling the nucleoside pocket. FicD variants previously appeared as biochemically indistinguishable, yet lead to different human pathologies. We demonstrate farnesyl-pyrophosphate inhibits FicDR374H and FicDR374C variants implicated in causing hereditary spastic paraplegia, but not the FicDR371S variant associated with neonatal diabetes. This study furthers our understanding of FicD inhibitors and distinguishes disease causing variants, providing insight into pharmacological targeting of UPR activity.
Beyond mobility: A prospective study on diet and metabolism in hereditary spastic paraplegia.
Metabolism plays an important role in neurodegenerative diseases. Hereditary spastic paraplegias (HSP) are a heterogeneous group of rare genetic neurodegenerative disorders, commonly characterized by the clinical syndrome of progressive lower limb spasticity and mobility loss. Obesity has been linked to distinct genotypes, but the role of metabolism and nutrition in HSP remains unclear.We aimed to To evaluate metabolism and nutrition in specific HSP genotypes and to assess the impact of nutritional counseling on disease progression and body composition. In this prospective explorative pilot study, we assessed the neurological, metabolic and nutritional status of patients with HSP at baseline and one year after nutritional counseling. A total of 36 patients with genetically confirmed SPG4-, SPG7- and SPG11-associated HSP were recruited. BMI in SPG4 and SPG7 was comparable to healthy population data, whereas SPG11 showed significantly higher BMI (+ 22.9%, p < 0.05) with a considerable interindividual variability. Across all genotypes, disease severity according to the Spastic Paraplegia Rating Scale correlated negatively with leg muscle mass (ρ = -0.39, p < 0.05), protein (ρ = -0.35, p < 0.05) and fiber intake (ρ = -0.41, p < 0.05). After one year, there was a significant loss of relative muscle mass (-7.2%, p < 0.001). Progressive loss of muscle mass in HSP asks for an effective nutritional intervention combined with exercise in order to influence disease progression in HSP. The SPG11-associated obese phenotype may evolve with disease progression due to multifactorial metabolic changes, beyond reduced mobility.
Modeling spastic paraplegia 4 with corticospinal motor neuron-enriched cortical organoids reveals genotype-phenotype and HDAC6-targetable pathology.
Spastic paraplegia 4 (SPG4), the most common form of hereditary spastic paraplegia, causes progressive gait deficiency due to corticospinal tract degeneration. SPG4 results from mutations in the SPAST gene, which encodes spastin, a microtubule-severing AAA-ATPase. To dissect genotype-phenotype relationships, we generated isogenic human induced pluripotent stem cell lines carrying either an SPAST missense (SPASTWT/C448Y) or truncation (SPASTWT/S245X) mutation and differentiated them into corticospinal motor neuron-enriched cortical organoids. These models revealed mutation-specific patterns of aberrant neuronal activity, microtubule hypoacetylation, and axonal degeneration. We identified mutant M1-spastin-induced hyperactivation of histone deacetylase 6 (HDAC6), a major tubulin deacetylase, as the key pathogenic culprit. Pharmacological inhibition of HDAC6 with tubastatin A restored microtubule acetylation and rescued axonal degeneration in organoids, with corresponding improvements in corticospinal tract integrity and gait defects in SPG4 transgenic mice. Our study uncovers HDAC6 hyperactivation as a targetable mechanism for SPG4 and verifies human organoids as a platform for therapeutic discovery.
Loss of function variants in HPDL impair human cortical development via alterations of mitochondrial function.
Human brain development is highly regulated by several spatiotemporal processes, which disruption can result in severe neurological disorders. Emerging evidence highlights the pivotal role of mitochondrial function as one of these fundamental pathways involved in neurodevelopment. Our study investigates the role of 4-hydroxyphenylpyruvate dioxygenase-like (HPDL) protein in cortical neurogenesis and mitochondrial activity, since mutations in the HPDL gene are associated with a childhood-onset form of hereditary spastic paraplegia characterized by corticospinal tract degeneration and cortical abnormalities. Starting from mutant neuroblastoma cells, we demonstrated that HPDL is important to respiratory chain supercomplex assembly and cellular redox balance. Moreover, RNA-seq studies revealed dysregulated pathways related to brain development. Generation of cortical neurons and organoids from HPDL patient-derived induced pluripotent stem cells exhibited premature neurogenesis at early differentiation stages, likely leading to depletion of cortical progenitors, as evidenced by decreased proliferation, slight increase of apoptosis, and unbalanced cortical type composition at later stages. Cortical organoids showed failure to grow at a normal rate, a feature highly reminiscent of the "microcephaly" observed in severe HPDL children. Mitochondrial morpho-functional characterization in mutant neurons confirmed disruption of OxPhos chain functionality in neuroblastoma knock-out model cells and HPDL mutant cortical progenitors also displayed defects in respirasome assembly and increased ROS generation rate. Treatment of mutant cortical cells with antioxidants and CoQ10 intermediates partially rescued premature neurogenesis depending on the mutational context, suggesting potential future personalized therapeutic strategies. Our findings reveal a critical role for HPDL in coordinating cortical progenitor proliferation, neurogenesis, and mitochondrial function, shedding light on a better understanding of the related clinical presentations.
MK4 Repositioning for IAHSP: Overcoming In Vivo Data Gaps through In Silico Refinement and In Vitro Validation.
Infantile-onset Ascending Hereditary Spastic Paralysis (IAHSP) is an ultrarare, autosomal recessive form of Hereditary Spastic Paraplegia (HSP), caused by mutations in the ALS2 gene, which encodes the protein ALSIN. In a previous study, we proposed a personalized therapeutic strategy for an Italian IAHSP patient (AO), aiming to correct the aberrant function of the R1611W mutant ALSIN using Menatetrenone (MK4). While our results supported compassionate-use approval for a patient-specific therapeutic regimen, further investigation was needed to highlight the treatment's benefits in the absence of tractable biophysical assays and in vivo models. In this respect, we first characterized MK4's interaction with the mutation site through Molecular Dynamics simulations. Next, we established and characterized a skin fibroblast cell line derived from patient AO. We analyzed the expression and stability of the mutant ALSIN protein in AO's fibroblasts and observed elevated oxidative stress levels. Using advanced microscopy and automated image analysis, we identified a characteristic mitochondrial phenotype associated with AO's IAHSP. One specific morphological parameter of mitochondria (Mean Branch Diameter) accurately reflected the IAHSP phenotype and was selected as a cell marker. Treatment of IAHSP fibroblasts with MK4 highlighted the rescue of Mean Branch Diameter and ALSIN levels, supporting cellular efficacy. Overall, this work presents an approach that integrates computational and cell-based methodologies to overcome the data scarcity challenges of drug discovery in rare diseases. Our study provides a framework for preclinical, alternative drug discovery programs in monogenic rare disorders such as IAHSP.
Publicações recentes
Expanding the clinical and mutational spectrum of hereditary spastic paraplegia type 4 in a cohort of patients from central China.
🥇 Meta-análiseSelective Dorsal Rhizotomy in Children with Hereditary Spastic Paraplegia.
🥇 Meta-análiseER discontinuities are common in C. elegans neurons, revealing a genetically tractable model for ER network maintenance.
Lipid droplets in neurodegenerative diseases: pathological drivers and therapeutic vulnerabilities.
📚 EuropePMC1.238 artigos no totalmostrando 197
Digenic inheritance of mutations in SPG7 and AFG3L2 causes motor neuron and cerebellar disorders.
BMC medicineX-linked adrenoleukodystrophy as an etiological cause of progressive spastic paraplegia: A case report.
The Journal of international medical researchAn MSA-P patient presenting with preserved glucose metabolism in the putamen, cerebellar hypometabolism and pronounced loss of presynaptic dopamine transporter in the striatum.
EJNMMI reportsFunctional validation of the novel KIF5A p.R17Q VUS reveals defective axonal transport in iPSC-motoneurons from a SPG10 patient.
Frontiers in geneticsRepetitive Transcranial Magnetic Stimulation for Spasticity in Stroke and Other Neuromotor Disorders: A Systematic Review of Randomized Clinical Trials.
Journal of clinical medicineFetal and Perinatal Brain MRI Findings in Adaptor Protein Complex 4-Associated Hereditary Spastic Paraplegia.
Annals of the Child Neurology SocietyPhosphatidylethanolamine: Its biological significance and responses to nutritional factors for neurohealth.
The Journal of nutritionSpastin Is Required to Prevent SPAST-Related Demyelination.
Journal of neurochemistryInhibition of FicD-mediated AMPylation and deAMPylation by isoprenoid diphosphates.
Proceedings of the National Academy of Sciences of the United States of AmericaBeyond mobility: A prospective study on diet and metabolism in hereditary spastic paraplegia.
Metabolic brain diseaseBeyond motor impairment: investigating cognitive aspects of hereditary spastic paraplegia in a Greek case series.
NeurocaseChildren with suspected hereditary spastic paraplegia clearly benefit from whole exome analysis.
Human genomicsA GJA1 Variant Triggers Earlier SPG4 Onset by Destabilizing Deubiquitinase VCPIP1 to Lower SPASTIN Levels.
Movement disorders : official journal of the Movement Disorder SocietyModeling spastic paraplegia 4 with corticospinal motor neuron-enriched cortical organoids reveals genotype-phenotype and HDAC6-targetable pathology.
Cell reportsMDSGene Systematic Review of Common Forms of Dominant Hereditary Spastic Paraplegia: Novel Insights.
Movement disorders clinical practiceLoss of function variants in HPDL impair human cortical development via alterations of mitochondrial function.
Cell death & diseaseStructural analysis of the plant glycoside hydrolase family 116 glucosylceramidase AtGCD3 by cryogenic electron microscopy.
International journal of biological macromoleculesRehabilitation with Intrathecal Baclofen and Gait Training Using Hybrid Assistive Limb in a Patient with Hereditary Spastic Paraplegia: A Case Report.
Progress in rehabilitation medicineTwo pathogenetic intronic variants in SPG4/SPAST and expansion of the clinical presentation.
Gene[Analysis of a three-generation Chinese pedigree affected with Hereditary spastic paraplegia type 3A due to variant of ATL1 gene].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsMK4 Repositioning for IAHSP: Overcoming In Vivo Data Gaps through In Silico Refinement and In Vitro Validation.
ACS chemical neuroscienceIdentification of an additional deep intronic splice variant prompts critical evaluation of SPG7 inheritance.
NeurogeneticsPlasma neurofilament light chain in pediatric hereditary spastic paraplegia.
Journal of the neurological sciencesA multi-ancestry genetic reference for the Quebec population.
Nature communicationsC. elegans Spastin/spas-1 Is Required for Axon Regeneration and Maintenance.
eNeuroNovel KIF5A variant in a patient with early-onset levodopa-responsive Parkinson's syndrome.
BMJ case reportsA Novel AP4M1 Variant in an Iranian Child with Spastic Paraplegia 50: A Case Report and Molecular Docking Approach.
Iranian journal of medical sciencesApoptosis and motor deficits in SPG76 hereditary spastic paraplegia: Calpain 2 inhibition as therapeutic strategy.
Pharmacological researchCo-development and implementation of a group-based arm-crank exercise programme in the community for individuals with neurological impairments.
BMC sports science, medicine & rehabilitationThe genetics of autosomal recessive ALS: a review of the common forms and their phenotypes.
Amyotrophic lateral sclerosis & frontotemporal degenerationTFG p.G269V Mutation Disrupts Motor Neuron Function in iPSC-Derived Models via Wnt Signaling Dysregulation.
Journal of neurochemistryExpansion of the genetic and phenotypic spectrum of hereditary spastic paraplegia caused by ABHD16A gene variants: an integrated analysis based on novel variants and literature review.
Frontiers in pediatricsMultiple Mitochondrial Dysfunction Syndrome Caused by IBA57 Gene Mutation: A Case Report and Literature Review.
Molecular genetics & genomic medicineNovel MTHFR variants manifesting with hereditary spastic paraplegia and recurrent pulmonary embolism: a case report and literature review of adult-onset severe MTHFR deficiency.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyLate-Onset X-linked Adrenoleukodystrophy: A Rare Cause of Progressive Spastic Paraparesis.
CureusAssociation of spinal cord structure with cognition in hereditary spastic paraplegia type 5.
Frontiers in neurologySerendipitous Symptom Control: Allopurinol for Spasticity in a Case of SPG4-Linked Hereditary Spastic Paraplegia-A Case Report.
Clinical case reportsHereditary spastic paraplegia: from decades of therapy to future innovations.
Therapeutic advances in neurological disordersEffect of an evidence-based modified developmental physiotherapy intervention on muscle tone, motor functions, and trunk control in a child with hereditary spastic paraplegia type 47: A case report.
Physiotherapy theory and practiceSubclinical involvement of central nervous system structures other than motor or sensory tracts in SPG3A and SPG4 patients.
BMC neurologyPearls & Oy-sters: Hereditary Spastic Paraplegia Type 15 Presenting as Juvenile Onset Levodopa-Responsive Parkinsonism.
NeurologyIdentification of myokymia in adult-onset hereditary spastic paraplegia type 79A: Implications for the phenotypic spectrum.
eNeurologicalSciFampridine in Hereditary Spastic Paraplegia Type 4 With SPAST Variant c.683-2A>C: A Case Report.
The American journal of case reportsNeurodevelopmental disorders in childhood-onset hereditary spastic paraplegia type 7: a case series and review of literature.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyDiagnostic Utility of the ATG9A Ratio in AP-4-Associated Hereditary Spastic Paraplegia.
Annals of clinical and translational neurologyFinite Element-Based Biomechanical Evaluation of Patient-Specific Insoles for a Pediatric Patient with Hereditary Spastic Paraplegia Using the Taguchi Method.
Bioengineering (Basel, Switzerland)A Mutation in Vesicular Acetylcholine Transporter Increases Tubulin Acetylation Compromising Synaptic Vesicle Transport.
Journal of neurochemistryComprehensive Characterization of Spastic Paraplegia in Korean Patients: A Single-Center Experience over Two Decades.
Yonsei medical journalExpanding the clinical phenotype of DYNC1H1 -associated mutations: a Chinese family with autosomal dominant complex hereditary spastic paraplegia.
BMC medical genomicsProtrudin acts at ER-endosome contacts to promote KIF5-mediated endosomal tubule fission.
Neurobiology of diseaseReversing Autophagy Inhibition Ameliorates Neurodegeneration in Hereditary Spastic Paraplegia Caused by a Degradation-Resistant SPAST Mutation.
Movement disorders : official journal of the Movement Disorder SocietyA new variant in the UCHL1 gene supporting its implication in late-onset ataxia with optic atrophy.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology[Hereditary spastic paraplegia of type 11: towards therapeutic options].
Medecine sciences : M/SGenetic and clinical characterization of SPG10: a case series of novel pathogenic variants and phenotypic diversity.
Annals of medicine and surgery (2012)Rare variation in neurological disease genes and its role in multiple sclerosis mimicry and phenotype.
Genome medicineLongitudinal Dynamics of Plasma Neurofilament Light Chain in Hereditary Spastic Paraplegia Type 11 (HSP-SPG11) and Type 15 (HSP-ZFYVE26).
Movement disorders : official journal of the Movement Disorder SocietyDeciphering Spastic Ataxia: Clinical and Genetic Profiles.
Neurology. GeneticsAutonomic profiling in SPG11 hereditary spastic paraplegia.
Clinical autonomic research : official journal of the Clinical Autonomic Research SocietyNovel missense ALDH18A1 variant in a family with autosomal dominant spastic paraplegia.
Journal of neurologyNovel SPTAN1 variant mimicking hereditary spastic paraplegia (HSP).
BMJ case reportsSpectrum of Movement Disorders in Early-Onset Hereditary Spastic Paraplegia: A Study of 428 Cases.
Movement disorders : official journal of the Movement Disorder SocietyClinical characteristics and gene mutation analysis of a family with hereditary spastic paraplegia type 11: a case report.
Journal of medical case reportsTwo homozygous KIF1C variants in a Turkish family presenting with cerebellar dysfunction and spastic paraparesis with MRI findings.
Parkinsonism & related disordersLoss-of-Function Variants in CPT1C: No Support for a Causal Role in Hereditary Spastic Paraplegia.
Movement disorders : official journal of the Movement Disorder SocietyIntracerebroventricular SPAST-AAV9 gene therapy prevents manifestation of symptoms in a mouse model of SPG4 hereditary spastic paraplegia.
Molecular therapy : the journal of the American Society of Gene TherapyKIF1A-associated neurological disorders: therapeutic opportunities and challenges.
European journal of human genetics : EJHGSwiss Cheese Gene Is Important for Intestinal Barrier, Microbiome, and Lipid Metabolism Regulation in Drosophila Gut.
International journal of molecular sciencesInfantile-Onset Ascending Hereditary Spastic Paraplegia due to a Homozygous ALS2 Exons 24-25 Deletion: Expanding the Genotypic Spectrum.
American journal of medical genetics. Part AMolecular Insights into IAHSP: Influence of the R1611W Mutation on the VPS9 Domain of Alsin.
ACS omegaThe Genetic Landscape of Hereditary Spastic Paraplegia in Greece.
Clinical geneticsThe genetic architecture of primary lateral sclerosis in a cohort of Italian patients.
Journal of neurologyREEP1 Accumulation Disrupts ER Integrity and Drives Spinal Motoneuron Degeneration in Distal Hereditary Motor Neuropathy.
Advanced science (Weinheim, Baden-Wurttemberg, Germany)DDHD2 possesses both lipase and transacylase capacities that remodel triglyceride acyl chains.
Proceedings of the National Academy of Sciences of the United States of AmericaMicrostructural Brain White Matter Changes and Clinical Correlates in SPG3A.
Movement disorders clinical practiceExpanding the Phenotypic Spectrum of ERLIN1-Related SPG62: Report of Two Siblings With Behavioral Features and Hyperacusis.
Clinical geneticsDorsolateral Cervical Cord T2 Hyperintensity in KIF1C-Related Disease (Spastic Paraplegia 58): Two Long-Duration Cases.
Annals of clinical and translational neurology[Clinical characteristics and genetic study of a child with Spastic paraplegia 52 due to variant of AP4S1 gene and a literature review].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsKyphoscoliosis peptidase deficiency-induced myofibrillar degeneration, focal depletion of mitochondria, and protein aggregation: A true myofibrillar myopathy?
Journal of neuromuscular diseasesERLIN1: A central regulator of protein quality control, lipid homeostasis, and cellular signaling at the endoplasmic reticulum.
Cellular signallingHealth-Related Quality of Life in Rare Forms of Childhood-Onset Hereditary Spastic Paraplegia.
Annals of clinical and translational neurologyMild cognitive dysfunction in hereditary spastic paraplegia 4 disease related to fluorodesoxyglucose cerebral positron emission tomography.
Brain communicationsArtificial intelligence in genomics: transforming the diagnosis of hereditary spastic paraplegia.
Annals of medicine and surgery (2012)Role of Oxidative Stress in Human Neurodegenerative Pathologies: Lessons from the Drosophila Model.
Current topics in medicinal chemistryA novel KIDINS220 mutation associated with hereditary spastic paraplegia accompanied by severe peripheral neuropathy.
Frontiers in neuroscienceChildren with Genetically Confirmed Hereditary Spastic Paraplegia: A Single-Center Experience.
Children (Basel, Switzerland)Fluid Biomarkers in Hereditary Spastic Paraplegia: A Narrative Review and Integrative Framework for Complex Neurodegenerative Mechanisms.
GenesBotulinum Toxin Treatment in Hereditary Spastic Paraplegia-A Comprehensive Review and Update.
ToxinsThe Neuro-Ophthalmologic Manifestations of SPG7-Associated Disease.
Journal of personalized medicineA Novel Frameshift Variant in the SPAST Gene Causing Hereditary Spastic Paraplegia in a Bulgarian-Turkish Family.
Neurology internationalHyperactivity of the non-canonical inflammasome in SPG11 and SPG48.
EBioMedicineEndoplasmic reticulum in the axon: Insights into structural dynamics and implications in neurodegeneration.
Neurobiology of diseaseTubular ER dysfunction in neurodegenerative diseases.
Neurobiology of disease4-Phenylbutyric Acid Improves Gait Ability of UBAP1-Related Spastic Paraplegia Mouse Model: Therapeutic Potential for SPG80.
International journal of molecular sciencesSPG7-Associated Ataxia May Involve Peripheral Neuropathy as a Key Phenotypic Feature.
Internal medicine (Tokyo, Japan)Hereditary Spastic Paraplegy Associated with the AP4S1 Gene: A Case Series Highlighting Diagnostic Pitfalls and Phenotypic Variability.
Molecular syndromologyDevelopment of a Human iPSC-Derived "Corticospinal Tract-on-a-Chip" for Neurodegenerative Disease Research.
bioRxiv : the preprint server for biologyThe cognitive profile of hereditary spastic paraplegia: a systematic review of the literature.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyDDHD2 provides a flux of saturated fatty acids for neuronal energy and function.
Nature metabolismA Case of Hereditary Spastic Paraplegia Type 50 With a Novel AP4M1 Variant and a Brief Review of the Literature.
Clinical case reportsAn atypical case of FA2H-related HSP35 with subtle neuroimaging findings and a novel variant in a young adult with spastic paraparesis.
Acta neurologica BelgicaWings of Discovery: Using Drosophila to Decode Hereditary Spastic Paraplegia and Ataxias.
CellsDysregulation of SELENOI Is Associated with TDP-43 Neuropathology in Amyotrophic Lateral Sclerosis.
CellsGenetic analysis of a family with skeletal muscle ion channelopathy and hereditary spastic paraplegia type 7 caused by SCN4A and SPG7 double mutations.
GeneGenetic and Clinical Investigations of C12orf65 Gene Mutations in Three Chinese Pedigrees.
Journal of neuro-ophthalmology : the official journal of the North American Neuro-Ophthalmology SocietyZiclague® (Alpinia Zerumbet oil) in patients with hereditary spastic paraplegia - the randomized controlled ZISPAST trial.
Orphanet journal of rare diseasesSerum NfL, but not GFAP, differentiates primary lateral sclerosis from adrenomyeloneuropathy and hereditary spastic paraplegia type 4.
Amyotrophic lateral sclerosis & frontotemporal degenerationEvidence of Dual Molecular Diagnosis of a Young Male Patient With Hereditary Spastic Paraplegia Carrying Two Rare Autosomal Mutations.
CureusPathogenic KIF1A R350 Variants Disrupt A Conserved Kinesin-Tubulin Salt Bridge.
bioRxiv : the preprint server for biologyExpanding the Phenotypic Spectrum of SPG4: Autism Spectrum Disorder in Early-Onset and Complex SPAST-HSP and Case Study.
GenesThe Impact of VAMP1 Mutations on Synaptic Vesicle Fusion Dynamics in Familial Spastic Disorders.
Journal of child neurologyA founder variant in TBCB is associated with global developmental delay, autism spectrum, and spastic paraparesis.
Genetics in medicine : official journal of the American College of Medical GeneticsWhole genome sequencing analysis in primary lateral sclerosis (PLS) patients reveals mutations in neurological diseases-causing genes.
Journal of neurology[A case of hereditary spastic paraplegia type 64 with ENTPD1 gene variant].
Zhonghua er ke za zhi = Chinese journal of pediatricsThe hereditary spastic paraplegia type 21 (SPG21) protein is a RAB7A effector that promotes noncanonical mTORC1-catalyzed TFEB phosphorylation and cytoplasmic retention.
Molecular biology of the cellLong-Term Clinical Characterization of ENTPD1-Related Spastic Paraplegia: A Novel Variant and Comprehensive Literature Review.
International journal of developmental neuroscience : the official journal of the International Society for Developmental NeuroscienceSPG7 p.A510V heterozygosity as a cause of adult-onset cerebellar ataxia without spasticity: longitudinal evidence from a sporadic case.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyNeuroaxonal Degeneration as a Converging Mechanism in Motor Neuron Diseases (MNDs): Molecular Insights into RNA Dysregulation and Emerging Therapeutic Targets.
International journal of molecular sciencesEfficacy of radial extracorporeal shock wave therapy in hereditary spastic paraplegia: A case report.
MedicineKIF1C-related disorders: spastic ataxia or hypomyelinating leukodystrophy? A paradigm of classification ambiguity.
NeurogeneticsGenotype-Phenotype Distinctions in Spastic Paraplegia 4 Reveal HDAC6 as a Therapeutic Target.
bioRxiv : the preprint server for biologyCharting the genetic landscape of autosomal recessive hereditary spastic paraplegia: A deep dive into 10 exceptionally rare cases.
NeurogeneticsSpinal cord structural changes in SPG4: insights from a large cohort using advanced neuroimaging.
Journal of neurologyDownbeat nystagmus in hereditary spastic paraplegia type 6.
Journal of neurologyWhen ganglioside pathways go awry: congenital disorders and experimental insights.
Journal of human geneticsAAV8-based gene replacement therapy for hereditary spastic paraplegia type 5.
Molecular therapy. Methods & clinical developmentYield of Whole Exome Sequencing in Children With Cryptogenic Cerebral Palsy.
Pediatric neurologyHPDL Biallelic Variants in Cerebral Palsy and Childhood-Onset Hereditary Spastic Paraplegia: Human and Zebrafish Insights.
Movement disorders : official journal of the Movement Disorder SocietyHereditary Spastic Paraplegia Type 7 With Early-Onset Parkinsonism Responsive to Subthalamic Deep Brain Stimulation.
Annals of Indian Academy of NeurologyEntropy, Irreversibility, and Time-Series Deep Learning of Kinematic and Kinetic Data for Gait Classification in Children with Cerebral Palsy, Idiopathic Toe Walking, and Hereditary Spastic Paraplegia.
Sensors (Basel, Switzerland)Adaptive Torque Control of Exoskeletons Under Spasticity Conditions via Reinforcement Learning.
IEEE ... International Conference on Rehabilitation Robotics : [proceedings]Implications of mitochondrial phosphatidylethanolamine in neuronal health and neurodegeneration.
Neural regeneration researchThe epidemiology of hereditary spastic paraplegia and associated common mental health outcomes in England and Northern Ireland.
Orphanet journal of rare diseasesTriglycerides are an important fuel reserve for synapse function in the brain.
Nature metabolismConformational dynamics and membrane insertion mechanism of B4GALNT1 in ganglioside synthesis.
Nature communicationsConnecting tubules: mechanisms of endoplasmic reticulum membrane fusion.
Biochemical Society transactionsHomozygous COQ9 mutation: a new cause of potentially treatable hereditary spastic paraplegia.
European journal of human genetics : EJHGNew variants and genotype-phenotype correlation in KIF5A mutation: the contribution of a large Italian cohort.
Journal of medical geneticsMaternal uniparental isodisomy in a patient with autosomal recessive spastic paraplegia type 20.
GeneNaringenin and SMER28 target lysosomal reformation and rescue SPG11 and SPG15 hereditary spastic paraplegia phenotypes.
Pharmacological researchExploring gastrocnemius medialis behavior during gait in children with cerebral palsy across different gait patterns.
Clinical biomechanics (Bristol, Avon)Twenty years of misdiagnosis of X-linked adrenoleukodystrophy: a case report.
American journal of translational researchGenotype variability in early-onset Hereditary Spastic Paraplegia: a single-center study.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyOne gene, many phenotypes: the role of KIF5A in neurodegenerative and neurodevelopmental diseases.
Cell communication and signaling : CCSIntrafamilial Phenotypic Variation in Taiwanese Patients with Hereditary Spastic Paraplegia and Charcot-Marie-Tooth Disease Due to KIF5A Mutations: A Cross-Sectional Observational Study.
Acta neurologica TaiwanicaBurden of pathogenetic and likely pathogenetic variants in SPG7, SPG11 and AP4 genes in Amyotrophic Lateral Sclerosis. A case-control study.
Journal of neurologyUnravelling axonal transcriptional landscapes: insights from induced pluripotent stem cell-derived cortical neurons and implications for motor neuron degeneration.
Open biologyOutcome measures of instrumented gait analysis in hereditary spastic paraplegia: a systematic review.
Journal of neuroengineering and rehabilitationFrench guidelines for the diagnosis and management of pure hereditary spastic paraplegia.
Revue neurologiqueStructural brain changes contributing to motor signs in pure hereditary spastic paraplegia type 4.
Journal of neurologyCase Report: Hereditary spastic paraplegia associated with monoallelic variant in the motor domain of KIF1A.
Frontiers in human neuroscienceA Japanese patient with hereditary spastic paraplegia with a rare KIF5A nonsense variant.
Human genome variationGenetic Evaluation of Patients with Clinically Suspected Hereditary Spastic Paraplegia with Seven Novel Variants.
Annals of Indian Academy of NeurologyExotropia in a Patient With a Novel Homozygous 4-Hydroxyphenylpyruvate Dioxygenase-Like Protein (HPDL) Variant.
CureusA Rare Homozygous AP4S1 Variant in Rwandan Siblings with Autosomal Recessive Hereditary Spastic Paraplegia Type 52 (SPG52).
GenesFrequently observed polyneuropathy expands phenotypic spectrum of apparently pure autosomal dominant hereditary spastic paraplegias.
Neurologia i neurochirurgia polskaGeneration of an induced pluripotent stem cell (iPSC) line carrying a KCNA2 homozygous (p.Arg294His, R294H) mutation related to hereditary spastic paraplegia.
Stem cell researchPathway Analyses of Inherited Neuropathies Identify Putative Common Mechanisms of Axon Degeneration.
Annals of clinical and translational neurologyKeratoconus in hereditary spastic paraplegia 15 and Kjellin syndrome: a case report.
Ophthalmic geneticsPhenotypic and molecular characterization of a recurrent SPTAN1 mutation causing SPG91.
Molecular biology reportsPrimary Lateral Sclerosis: Implications for Diagnostic Criteria From a Natural History Study in the Netherlands.
NeurologySpastic paraplegia with short stature: Think of Troyer syndrome.
Acta neurologica BelgicaDiagnostic journey and genetic analysis of a novel homozygous CYP2U1 mutation causing autosomal recessive spastic paraplegia type 56 (SPG56) in a consanguineous family.
BMC neurologyHPDL Variant Type Correlates With Clinical Disease Onset and Severity.
Annals of clinical and translational neurologyA decade of whole-exome sequencing in Brazilian Neurology: from past insights to future perspectives.
Arquivos de neuro-psiquiatria[Genetics of Motor Neuron Diseases and Hereditary Spastic Paraplegia].
Brain and nerve = Shinkei kenkyu no shinpoHereditary Spastic Paraplegia in Alberta: Lessons from a Well-Defined Cohort Including the Indigenous Population.
Movement disorders clinical practiceKIF5A variant in familial dystonia: A clinicogenetic study of a large Roma kindred.
Parkinsonism & related disordersExpanding the Allelic and Clinical Heterogeneity of Movement Disorders Linked to Defects of Mitochondrial Adenosine Triphosphate Synthase.
Movement disorders : official journal of the Movement Disorder SocietyUCHL1-Mediated Spastin Degradation Regulates Microtubule Severing and Hippocampal Neurite Outgrowth.
Journal of molecular neuroscience : MNap4b1 -/- zebrafish demonstrate morphological and motor abnormalities.
Human molecular geneticsMicroglia and CD8+ T cell activation precede neuronal loss in a murine model of spastic paraplegia 15.
The Journal of experimental medicineImpact of pathogenic mutations on the refolding ability and stability of human mitochondrial Phenylalanyl-tRNA synthetase.
Archives of biochemistry and biophysicsLipid Droplet in Lipodystrophy and Neurodegeneration.
Biology of the cellArginase 1 deficiency: a treatable form of spastic paraplegia.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyGait Training for Walking Acquisition in a Child with Hereditary Spastic Paraplegia: A Case Report.
Pediatric physical therapy : the official publication of the Section on Pediatrics of the American Physical Therapy AssociationIncidence and health burden of 20 rare neurological diseases in South China from 2016 to 2022: a hospital-based observational study.
Orphanet journal of rare diseasesPathogenic mutation impairs functional dynamics of Hsp60 in mono- and oligomeric states.
Nature communicationsExpanding the Clinical Spectrum of SPG26: A Case Report and Review of B4GALNT1-Associated Hereditary Spastic Paraplegia.
Movement disorders clinical practicePatient-specific mutation of contact site protein Tomm70 causes neurodegeneration.
Disease models & mechanismsSensory Dysfunction in ALS and Other Motor Neuron Diseases: Clinical Relevance, Histopathology, Neurophysiology, and Insights from Neuroimaging.
BiomedicinesInsights from selenoprotein I mouse models for understanding biological roles of this enzyme.
Archives of biochemistry and biophysicsToxoplasmosis accelerates the progression of hereditary spastic paraplegia.
mSphereHereditary spastic paraplegia HSP26: clinical and electrophysiological characteristics.
Neurophysiologie clinique = Clinical neurophysiologyFIC Domain Protein Adenylyltransferase (FICD)-Related Complex Hereditary Spastic Paraplegia with Diabetes Mellitus.
Movement disorders clinical practiceFlexibility, Resistance, Aerobic, Movement Execution (FRAME) training program to improve gait capacity in adults with Hereditary Spastic Paraplegia: protocol for a single-cohort feasibility trial.
Frontiers in neurologyMotor Neuron Involvement in Two ATP13A2-Related Families: ALS And HSP-Like Phenotypes.
Movement disorders clinical practiceArrayed CRISPR/Cas9 Loss-Of-Function Screen in a Neuronal Model of Adaptor Protein Complex 4 Deficiency Identifies Modulators of ATG9A Trafficking.
bioRxiv : the preprint server for biologyNovel SPAST Deletion Mutation in an American Family With Hereditary Spastic Paraplegia: A Case Report.
Journal of investigative medicine high impact case reportsTargeted Long-Read Sequencing as a Single Assay Improves the Diagnosis of Spastic-Ataxia Disorders.
Annals of clinical and translational neurologyNovel Aspects of Hereditary Spastic Paraplegia: A Clinicopathologic and Biochemical Study of a Patient With a Heterozygous GCH1 Variant.
Neuropathology and applied neurobiologyThe Spastic Paraplegia-Centers of Excellence Research Network (SP-CERN): Clinical Trial Readiness for Hereditary Spastic Paraplegia.
Neurology. GeneticsPartial loss of FITM2 function causes hereditary spastic paraplegia.
medRxiv : the preprint server for health sciencesIf at first you don't succeed, try, try again.
Survey of ophthalmologyNeurogenic arthrogryposis, hypotonia, dysmorphic features plus malformation of cortical development further expands the ARL6IP1 loss-of-function phenotype.
Neuromuscular disorders : NMDSpastic Paraplegia Type 78 Associated With ATP13A2 Gene Variants in Compound Heterozygosity.
Molecular genetics & genomic medicineOculodentodigital Dysplasia Presenting as Spastic Ataxic Syndrome in an Indian Patient.
Annals of Indian Academy of NeurologyNeurodevelopmental Implications Underpinning Hereditary Spastic Paraplegia.
CNS neuroscience & therapeuticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Paraplegia espástica hereditária.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Paraplegia espástica hereditária
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Inhibition of FicD-mediated AMPylation and deAMPylation by isoprenoid diphosphates.Proceedings of the National Academy of Sciences of the United States of America· 2026· PMID 41779785mais citado
- Beyond mobility: A prospective study on diet and metabolism in hereditary spastic paraplegia.
- Modeling spastic paraplegia 4 with corticospinal motor neuron-enriched cortical organoids reveals genotype-phenotype and HDAC6-targetable pathology.
- Loss of function variants in HPDL impair human cortical development via alterations of mitochondrial function.
- MK4 Repositioning for IAHSP: Overcoming In Vivo Data Gaps through In Silico Refinement and In Vitro Validation.
- Expanding the clinical and mutational spectrum of hereditary spastic paraplegia type 4 in a cohort of patients from central China.
- Correction to "Loss-of-Function Variants in CPT1C: No Support for a Causal Role in Hereditary Spastic Paraplegia".
- Selective Dorsal Rhizotomy in Children with Hereditary Spastic Paraplegia.
- ER discontinuities are common in C. elegans neurons, revealing a genetically tractable model for ER network maintenance.
- Lipid droplets in neurodegenerative diseases: pathological drivers and therapeutic vulnerabilities.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:685(Orphanet)
- MONDO:0019064(MONDO)
- GARD:6637(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q657516(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar