Qualquer condição em que o feto apresenta um conjunto de problemas de formação e alterações físicas, causados pela falta de movimento, e cuja causa é uma alteração (mutação) no gene MUSK.
Introdução
O que você precisa saber de cara
Qualquer condição em que o feto apresenta um conjunto de problemas de formação e alterações físicas, causados pela falta de movimento, e cuja causa é uma alteração (mutação) no gene MUSK.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 30 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 63 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
10 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisLigand for NRCAM and NFASC/neurofascin that plays a role in the formation and maintenance of the nodes of Ranvier on myelinated axons. Mediates interaction between Schwann cell microvilli and axons via its interactions with NRCAM and NFASC. Nodes of Ranvier contain clustered sodium channels that are crucial for the saltatory propagation of action potentials along myelinated axons. During development, nodes of Ranvier are formed by the fusion of two heminodes. Required for normal clustering of so
Cell membraneCell projection, axonSecretedSecreted, extracellular space, extracellular matrix
Lethal congenital contracture syndrome 11
A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy and congenital non-progressive joint contractures. The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth.
Acts as a transcriptional activator that promotes transcription of muscle-specific target genes and plays a role in muscle differentiation. Together with MYF5 and MYOG, co-occupies muscle-specific gene promoter core region during myogenesis. Induces fibroblasts to differentiate into myoblasts. Interacts with and is inhibited by the twist protein. This interaction probably involves the basic domains of both proteins (By similarity)
Nucleus
Congenital myopathy 17
An autosomal recessive muscular disorder characterized by hypotonia and respiratory insufficiency apparent soon after birth, high diaphragmatic dome on imaging, poor overall growth, pectus excavatum, dysmorphic facies, and renal anomalies in some affected individuals. Additional variable features include delayed motor development, mildly decreased endurance, distal arthrogryposis, and lung hypoplasia resulting in early death.
Processive microtubule plus-end directed motor protein involved in neuronal axon guidance. Is recruited by KANK1 to cortical microtubule stabilizing complexes (CMSCs) at focal adhesions (FAs) rims where it promotes microtubule capture and stability. Controls microtubule polymerization rate at axonal growth cones and suppresses microtubule growth without inducing microtubule disassembly once it reaches the cell cortex
Cytoplasm, cytoskeletonCytoplasm, cell cortexCell projection, axonCell projection, dendriteCell projection, growth cone
Fibrosis of extraocular muscles, congenital, 1
A congenital ocular motility disorder marked by restrictive ophthalmoplegia affecting extraocular muscles innervated by the oculomotor and/or trochlear nerves. It is clinically characterized by anchoring of the eyes in downward gaze, ptosis, and backward tilt of the head. Patients affected by congenital fibrosis of extraocular muscles type 1 show an absence of the superior division of the oculomotor nerve (cranial nerve III) and corresponding oculomotor subnuclei.
Component of nuclear pore complex
Nucleus, nuclear pore complex
Fetal akinesia deformation sequence 4
A clinically and genetically heterogeneous group of disorders with congenital malformations related to impaired fetal movement. Clinical features include fetal akinesia, intrauterine growth retardation, polyhydramnios, arthrogryposis, pulmonary hypoplasia, craniofacial abnormalities, and cryptorchidism. FADS4 inheritance is autosomal recessive.
Postsynaptic protein required for clustering of nicotinic acetylcholine receptors (nAChRs) at the neuromuscular junction. It may link the receptor to the underlying postsynaptic cytoskeleton, possibly by direct association with actin or spectrin
Cell membranePostsynaptic cell membraneCytoplasm, cytoskeleton
Myasthenic syndrome, congenital, 11, associated with acetylcholine receptor deficiency
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS11 is an autosomal recessive disorder of postsynaptic neuromuscular transmission, due to deficiency of AChR at the endplate that results in low amplitude of the miniature endplate potential and current.
Electrogenic antiporter that exchanges one cholinergic neurotransmitter, acetylcholine or choline, with two intravesicular protons across the membrane of synaptic vesicles. Uses the electrochemical proton gradient established by the V-type proton-pump ATPase to store neurotransmitters inside the vesicles prior to their release via exocytosis (By similarity) (PubMed:20225888, PubMed:8910293). Determines cholinergic vesicular quantal size at presynaptic nerve terminals in developing neuro-muscular
Cytoplasmic vesicle, secretory vesicle, synaptic vesicle membrane
Myasthenic syndrome, congenital, 21, presynaptic
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS21 is an autosomal recessive, pre-synaptic form characterized by ptosis, ophthalmoplegia, fatigable weakness, apneic crises, and deterioration of symptoms in cold water. Learning difficulties and left ventricular dysfunction may be present in some patients.
Probably enhances ubiquitin ligase activity of RING-type zinc finger-containing E3 ubiquitin-protein ligases, possibly through recruitment and/or stabilization of the Ubl-conjugating enzyme (E2) at the E3:substrate complex. Acts as a regulator of retrograde transport via its interaction with VPS35. Recruited to retromer-containing endosomes and promotes the formation of 'Lys-63'-linked polyubiquitin chains at 'Lys-220' of WASHC1 together with TRIM27, leading to promote endosomal F-actin assembly
Early endosomeCytoplasmNucleus
Schaaf-Yang syndrome
A disease characterized by clinical features of Prader-Willi syndrome, including neonatal hypotonia with poor suck, feeding problems in infancy, obesity, developmental delay, short stature, and hypogonadism. Additionally, patients manifest autism spectrum disorder. Some patients have dysmorphic facial features.
Probable muscle-intrinsic activator of MUSK that plays an essential role in neuromuscular synaptogenesis. Acts in aneural activation of MUSK and subsequent acetylcholine receptor (AchR) clustering in myotubes. Induces autophosphorylation of MUSK
Cell membraneSynapse
Myasthenic syndrome, congenital, 10
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS10 is an autosomal recessive, post-synaptic form characterized by a typical 'limb girdle' pattern of muscle weakness with small, simplified neuromuscular junctions but normal acetylcholine receptor and acetylcholinesterase function.
Tubulin is the major constituent of microtubules, a cylinder consisting of laterally associated linear protofilaments composed of alpha- and beta-tubulin heterodimers. Microtubules grow by the addition of GTP-tubulin dimers to the microtubule end, where a stabilizing cap forms. Below the cap, tubulin dimers are in GDP-bound state, owing to GTPase activity of alpha-tubulin
Cytoplasm, cytoskeletonCytoplasm, cytoskeleton, flagellum axoneme
Lissencephaly 3
A classic type lissencephaly associated with psychomotor retardation and seizures. Features include agyria or pachygyria or laminar heterotopia, severe intellectual disability, motor delay, variable presence of seizures, and abnormalities of corpus callosum, hippocampus, cerebellar vermis and brainstem.
Receptor tyrosine kinase which plays a central role in the formation and the maintenance of the neuromuscular junction (NMJ), the synapse between the motor neuron and the skeletal muscle (PubMed:25537362). Recruitment of AGRIN by LRP4 to the MUSK signaling complex induces phosphorylation and activation of MUSK, the kinase of the complex. The activation of MUSK in myotubes regulates the formation of NMJs through the regulation of different processes including the specific expression of genes in s
Postsynaptic cell membrane
Myasthenic syndrome, congenital, 9, associated with acetylcholine receptor deficiency
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS9 is a disorder of postsynaptic neuromuscular transmission, due to deficiency of AChR at the endplate that results in low amplitude of the miniature endplate potential and current.
Variantes genéticas (ClinVar)
115 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 2.544 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
63 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Sequência de deformação de acinesia fetal
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Perinatal genetic diagnostic yield in a population of fetuses with the phenotype arthrogryposis multiplex congenita: a cohort study 2007-2021.
Arthrogryposis multiplex congenita (AMC) presents challenges for prenatal detection due to its heterogeneous etiology, onset, and phenotypical manifestations. This study aims to describe the genetic diagnostic yield in a population of fetuses with detailed phenotypic description over a 15-year period (2007-2021) at the Fetal Medicine Unit of Amsterdam UMC, the Netherlands. The fetal and neonatal phenotypes were classified into three clinical AMC Groups, with the exception that Groups 1 and 2 were combined in the prenatal classification. Group 1 involves limb involvement primarily, Group 2 includes musculoskeletal involvement plus other system anomalies, and Group 3 involves musculoskeletal involvement with central nervous system disability, lethality, fetal akinesia deformation sequence, and/or intellectual disability. The cohort consisted of 64 consecutive cases, 13 in Groups 1 + 2 and 51 in Group 3. Perinatal genetic testing occurred in all cases: prenatally in 56 of the 64 (88%), postnatally in 36 of the 64 (56%), and combined testing in 28 of the 64 cases (44%). The overall genetic diagnostic yield was 28% (18/64), and it increased over the 5-year period from 14% to 50%. Whole exome sequencing had the highest yield (41.7%). The yield per phenotype was 30.8% (4/13) for AMC Group 1 + 2 and 27.4% (14/51) for AMC Group 3. Detailed fetal phenotyping and perinatal genetic testing in all cases showed improved diagnostic yield over time, likely due to the introduction of Next-generation sequencing-based tests. The availability of stored DNA will be beneficial for future investigations since further improvements in genetic testing possibilities are expected.
Whole Genome Sequence Identifies the Second Allele: An Intronic Variant in RYR1 Contributes to Early-Onset Fetal Akinesia Deformation Sequence.
Fetal akinesia deformation sequence (FADS), a severe prenatal phenotype associated with congenital myopathies (CMs), is linked to mutations in the RYR1 gene. Although whole exome sequencing (WES) is a standard diagnostic approach, certain pathogenic variants may remain undetected. In this study, we utilized whole genome sequencing (WGS) to investigate a recurrent case of FADS with negative WES results. A couple who experienced four consecutive pregnancy losses, including two fetuses affected by FADS, underwent trio-WGS following negative WES findings. Variants were annotated using public databases and filtered according to ACMG guidelines. Functional validation of an intronic variant was conducted using RT-PCR, TA cloning, and Sanger sequencing. WGS identified two novel RYR1 compound heterozygous variants in the affected fetus: a paternally inherited in-frame mutation (c.13659+655_14172 +588delinsCTGGCGCCCCATCTCAT) located the C-terminal hotspot and a maternally inherited deep intronic variant (c.4934+25G>A). Both of them were initially classified as variant of uncertain significance. RNA studies demonstrated that this intronic variant caused a 26-bp intron retention and frameshift, leading to its reclassification as pathogenic. And according to this PM3 evidence, the in-frame mutation was reclassified as likely pathogenic. Both variants were absent from population databases and exhibited segregation with the phenotype. This study highlights the utility of WGS in diagnosing fetal anomalies with negative WES results by identifying noncoding and structural variants. The identification of novel RYR1 mutations broadens the genetic spectrum of recessive CMs and emphasizes the necessity for functional assays to accurately interpret intronic variants. WGS is recommended for cases of recurrent fetal anomalies when WES is inconclusive, although careful interpretation of variants is advised.
Preemptive immunotherapy for fetal acetylcholine receptor antibody disorder (FARAD): from recurrent pregnancy losses to a healthy infant - a case report.
Maternal antibodies against the fetal acetylcholine receptor (fAChR) can cause fetal acetylcholine receptor antibody-related disorders (FARAD), a rare but important cause of arthrogryposis multiplex congenita (AMC) and Fetal Akinesia Deformation Sequence (FADS), with recurrence rates up to 100%. Unlike genetic causes, FARAD may respond to maternal immunotherapy, yet standardized treatment protocols are lacking. We report four pregnancies in an asymptomatic mother with confirmed fAChR antibodies. After three fetal losses due to FARAD and two unsuccessful treatment attempts initiated only after sonographic abnormalities, a healthy infant was delivered in the fourth pregnancy following a pre-planned preemptive approach. Treatment, initiated at week 10 of gestation, consisted of plasmapheresis (PLEX) and subsequent weekly high-dose intravenous immunoglobulin (IVIg). Fetal serum after birth confirmed fAChR antibodies on cell-based assays (live and fixed). This case series underscores the importance of recognizing fAChR antibodies as differential diagnosis of AMC in asymptomatic mothers and of early, consistent antibody-reducing therapy to prevent FARAD-related pregnancy losses.
Variant Update on ASCC1 : Characterization of the First Homozygous Missense Variant Involved in Prenatal-Onset Spinal Muscular Atrophy With Congenital Bone Fractures 2.
Spinal muscular atrophy with congenital bone fractures 2 is a rare and severe autosomal recessive neuromuscular disorder caused by pathogenic variants in ASCC1. This condition characterized by prenatal onset of severe hypotonia with fetal hypokinesia and congenital contractures results in arthrogryposis multiplex congenita, and increased incidence of prenatal fractures. To date, only truncating variants, loss of function and splicing variants have been described. Here, we report the first homozygous missense variant in ASCC1 identified prenatally in two full siblings with fetal akinesia deformation sequence. This variant affects a highly conserved residue within the RNA-ligase-like domain and leads to a nearly total absence of ASCC1 protein in muscle. This report broadens the knowledge on the pathogenesis of this disorder showing that missense variants should also be considered. It also highlights the importance of precise ultrasound examination combined with molecular genetic testing in the prenatal diagnosis of this severe neuromuscular disorder.
DOK7 Gene Novel Homozygous Mutation is Related to Fetal Akinesia Deformation Sequence 3.
Fetal akinesia deformation sequence syndrome with a prevalence of 1 per 13:000 refers to a clinically and genetically heterogeneous disorder recognized by joint contractures, pterygia, fetal hydrops, dysmorphic features and lung hypoplasia's common features. Both genetic and parental/external environmental factors can result in this syndrome. DOK7 mutations will result in Fetal akinesia deformation sequence 3; the inheritance pattern of the named gene is AR and its protein has a major role as a signaling molecule necessary for neuromuscular junction. In this study, a couple who had three recurrent abortions were referred to the Genome laboratory of Isfahan in Iran. Pathological, immunological and hormonal tests were requested for the mother in the first stage, and also Giemsa banding karyotype were requested for the father and mother. Next, array comparative genomic hybridization (array CGH) was requested for the aborted fetus sampling, and whole-exome sequencing was done to mutation analysis. Here, for the first time we report a case which contains novel homozygote mutation NM_173660:exon4:c.G481A:p.G161R in DOK7 gene locates on 4p16.3 as a novel mutation of the DOK7 gene that is a pathogenic variant and may play an important role in Fetal akinesia deformation sequence 3. Homozygote mutation NM_173660:exon4:c.G481A:p.G161R in DOK7 gene as a pathogenic variant may play an important role in Fetal akinesia deformation sequence 3 that directly results in recurring miscarriage.
Publicações recentes
Whole Genome Sequence Identifies the Second Allele: An Intronic Variant in RYR1 Contributes to Early-Onset Fetal Akinesia Deformation Sequence.
Preemptive immunotherapy for fetal acetylcholine receptor antibody disorder (FARAD): from recurrent pregnancy losses to a healthy infant - a case report.
Variant Update on ASCC1 : Characterization of the First Homozygous Missense Variant Involved in Prenatal-Onset Spinal Muscular Atrophy With Congenital Bone Fractures 2.
DOK7 Gene Novel Homozygous Mutation is Related to Fetal Akinesia Deformation Sequence 3.
The evolving genetic landscape of neuromuscular fetal akinesias.
📚 EuropePMC60 artigos no totalmostrando 65
Whole Genome Sequence Identifies the Second Allele: An Intronic Variant in RYR1 Contributes to Early-Onset Fetal Akinesia Deformation Sequence.
Molecular syndromologyPreemptive immunotherapy for fetal acetylcholine receptor antibody disorder (FARAD): from recurrent pregnancy losses to a healthy infant - a case report.
Neuromuscular disorders : NMDVariant Update on ASCC1 : Characterization of the First Homozygous Missense Variant Involved in Prenatal-Onset Spinal Muscular Atrophy With Congenital Bone Fractures 2.
American journal of medical genetics. Part ADOK7 Gene Novel Homozygous Mutation is Related to Fetal Akinesia Deformation Sequence 3.
Journal of obstetrics and gynaecology of IndiaThe evolving genetic landscape of neuromuscular fetal akinesias.
Journal of neuromuscular diseasesBiallelic Variant, c.644-13_644-9del in UNC50 Is Associated With Congenital Myasthenia Syndrome.
American journal of medical genetics. Part APerinatal genetic diagnostic yield in a population of fetuses with the phenotype arthrogryposis multiplex congenita: a cohort study 2007-2021.
European journal of human genetics : EJHGBiallelic variants in AGRN in a family with recurrent pregnancy losses and fetal akinesia deformation sequence.
Clinical dysmorphologyIdentification of a Founder GLDN Variant Associated With "Lethal" Arthrogryposis in Nunavik Inuit: Implications for Obstetrical and Long-Term Survivors' Management.
American journal of medical genetics. Part AThe p.R66W Variant in RAC3 Causes Severe Fetopathy Through Variant-Specific Mechanisms.
CellsECEL1 mutation in distal arthrogryposis type 5D: A case report.
European journal of obstetrics, gynecology, and reproductive biologyIdentification of four TTN variants in three families with fetal akinesia deformation sequence.
BMC medical genomicsNovel variant in ACTA1 identified in a fetus with akinesia deformation sequence and cortical development delay.
Prenatal diagnosisThe severity of MUSK pathogenic variants is predicted by the protein domain they disrupt.
HGG advancesClinical and molecular characteristics of 26 fetuses with lethal multiple congenital contractures.
Clinical geneticsAlteration of actin cytoskeletal organisation in fetal akinesia deformation sequence.
Scientific reportsBiallellic variants in CACNA1S cause fetal akinesia sequence, progressive hydrops and stillbirth.
Prenatal diagnosisSevere neuromuscular forms of glycogen storage disease type IV: Histological, clinical, biochemical, and molecular findings in a large French case series.
Journal of inherited metabolic diseaseVariants in DOK7 results in fetal akinesia deformation sequence: A case report and review of literature.
Clinical geneticsFetal akinesia deformation sequence syndrome associated with recessive TTN variants.
American journal of medical genetics. Part AClinically diverse and perinatally lethal syndromes with urorectal septum malformation sequence.
American journal of medical genetics. Part ARestrictive dermopathy: A baby with taut skin, facial dysmorphism, joint contractures, and pulmonary hypoplasia.
JAAD case reportsLethal Congenital Contracture Syndrome 11: A Case Report and Literature Review.
Journal of clinical medicineBroadening the phenotypic spectrum of TUBA1A tubulinopathy to syndromic arthrogryposis multiplex congenita.
American journal of medical genetics. Part AA single center experience of prenatal parent-fetus trio exome sequencing for pregnancies with congenital anomalies.
Prenatal diagnosisFetal akinesia deformation sequence and massive perivillous fibrin deposition resulting in fetal death in six fetuses from one consanguineous couple, including literature review.
Molecular genetics & genomic medicineFetal akinesia deformation sequence with pontocerebellar hypoplasia, and migration and gyration defects.
Autopsy & case reportsJoint development recovery on resumption of embryonic movement following paralysis.
Disease models & mechanismsDeformations associated with arthrogryposis.
American journal of medical genetics. Part ACHRNB1-associated congenital myasthenia syndrome: Expanding the clinical spectrum.
American journal of medical genetics. Part ACentrosome and ciliary abnormalities in fetal akinesia deformation sequence human fibroblasts.
Scientific reportsNeurogenetic fetal akinesia and arthrogryposis: genetics, expanding genotype-phenotypes and functional genomics.
Journal of medical geneticsThe latest FADS: Functional analysis of GLDN patient variants and classification of GLDN-associated AMC as a type of viable fetal akinesia deformation sequence.
American journal of medical genetics. Part ABiallelic c.1263dupC in DOK7 results in fetal akinesia deformation sequence.
American journal of medical genetics. Part APrenatal sonographic diagnosis of Dandy-Walker malformation and type III lissencephaly: A novel association.
Journal of clinical ultrasound : JCUCompound heterozygous mutation of MUSK causing fetal akinesia deformation sequence syndrome: A case report.
World journal of clinical casesNull variants in AGRN cause lethal fetal akinesia deformation sequence.
Clinical geneticsDiagnosis of fetal non-chromosomal abnormalities on routine ultrasound examination at 11-13 weeks' gestation.
Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and GynecologyFetal arthrogryposis multiplex congenita/fetal akinesia deformation sequence (FADS)-Aetiology, diagnosis, and management.
Prenatal diagnosisSevere biallelic loss-of-function mutations in nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) in two fetuses with fetal akinesia deformation sequence.
Experimental neurologySLC18A3 variants lead to fetal akinesia deformation sequence early in pregnancy.
American journal of medical genetics. Part ANext generation sequencing in recurrent pregnancy loss-approaches and outcomes.
European journal of medical geneticsIsolated vocal cord paralysis in two siblings with compound heterozygous variants in MUSK: Expanding the phenotypic spectrum.
American journal of medical genetics. Part AHomozygous/compound heterozygote RYR1 gene variants: Expanding the clinical spectrum.
American journal of medical genetics. Part AFetal akinesia deformation sequence, arthrogryposis multiplex congenita, and bilateral clubfeet: Is motor assessment of additional value for in utero diagnosis? A 10-year cohort study.
Prenatal diagnosisBiallelic mutations in nucleoporin NUP88 cause lethal fetal akinesia deformation sequence.
PLoS geneticsPena-Shokeir syndrome: current management strategies and palliative care.
The application of clinical geneticsNovel pathogenic variants in GBE1 causing fetal akinesia deformation sequence and severe neuromuscular form of glycogen storage disease type IV.
Clinical dysmorphologyMolecular autopsy by trio exome sequencing (ES) and postmortem examination in fetuses and neonates with prenatally identified structural anomalies.
Genetics in medicine : official journal of the American College of Medical GeneticsAppropriate use of next generation sequencing facilities for identifying new genetic causes of fetal akinesia deformation sequence (FADS).
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyMassive parallel sequencing identifies RAPSN and PDHA1 mutations causing fetal akinesia deformation sequence.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyLethal multiple pterygium syndrome: A severe phenotype associated with a novel mutation in the nebulin gene.
Neuromuscular disorders : NMDMutations in the NEB gene cause fetal akinesia/arthrogryposis multiplex congenita.
Prenatal diagnosisType 0 Spinal Muscular Atrophy: Further Delineation of Prenatal and Postnatal Features in 16 Patients.
Journal of neuromuscular diseasesIdentification of KLHL40 mutations by targeted next-generation sequencing facilitated a prenatal diagnosis in a family with three consecutive affected fetuses with fetal akinesia deformation sequence.
Prenatal diagnosisDiscordant monoamniotic twins with Pena-Shokeir phenotype.
Clinical case reportsCongenital Zika Virus Infection: Beyond Neonatal Microcephaly.
JAMA neurologyA novel neuromuscular form of glycogen storage disease type IV with arthrogryposis, spinal stiffness and rare polyglucosan bodies in muscle.
Neuromuscular disorders : NMDPrenatal diagnosis of fetal akinesia deformation sequence (FADS): a study of 79 consecutive cases.
Archives of gynecology and obstetricsDeficiency of the myogenic factor MyoD causes a perinatally lethal fetal akinesia.
Journal of medical geneticsDe novo TUBB2B mutation causes fetal akinesia deformation sequence with microlissencephaly: An unusual presentation of tubulinopathy.
European journal of medical geneticsExome sequencing positively identified relevant alterations in more than half of cases with an indication of prenatal ultrasound anomalies.
Prenatal diagnosisFetal akinesia deformation sequence due to a congenital disorder of glycosylation.
American journal of medical genetics. Part ATargeted carrier screening for four recessive disorders: high detection rate within a founder population.
European journal of medical geneticsMuSK: a new target for lethal fetal akinesia deformation sequence (FADS).
Journal of medical geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Sequência de deformação de acinesia fetal.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Sequência de deformação de acinesia fetal
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Perinatal genetic diagnostic yield in a population of fetuses with the phenotype arthrogryposis multiplex congenita: a cohort study 2007-2021.
- Whole Genome Sequence Identifies the Second Allele: An Intronic Variant in RYR1 Contributes to Early-Onset Fetal Akinesia Deformation Sequence.
- Preemptive immunotherapy for fetal acetylcholine receptor antibody disorder (FARAD): from recurrent pregnancy losses to a healthy infant - a case report.
- Variant Update on ASCC1 : Characterization of the First Homozygous Missense Variant Involved in Prenatal-Onset Spinal Muscular Atrophy With Congenital Bone Fractures 2.
- DOK7 Gene Novel Homozygous Mutation is Related to Fetal Akinesia Deformation Sequence 3.
- The evolving genetic landscape of neuromuscular fetal akinesias.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:994(Orphanet)
- OMIM OMIM:208150(OMIM)
- MONDO:0100101(MONDO)
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q54366512(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
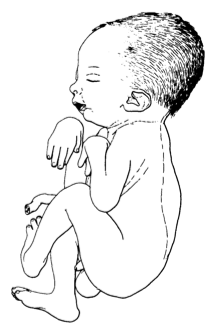
Sequência de deformação de acinesia fetal
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata