Transtorno neurogenético caracterizado por grave déficit intelectual e características dismórficas faciais distintas.
Introdução
O que você precisa saber de cara
Transtorno neurogenético caracterizado por grave déficit intelectual e características dismórficas faciais distintas.
Tem tratamento?
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 47 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 113 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
4 genes identificados com associação a esta condição. Padrão de herança: Not applicable.
Curadoria gene-doença
fontes oficiaisE3 ubiquitin-protein ligase which accepts ubiquitin from an E2 ubiquitin-conjugating enzyme in the form of a thioester and transfers it to its substrates (PubMed:10373495, PubMed:16772533, PubMed:19204938, PubMed:19233847, PubMed:19325566, PubMed:19591933, PubMed:22645313, PubMed:24273172, PubMed:24728990, PubMed:30020076). Several substrates have been identified including the BMAL1, ARC, LAMTOR1, RAD23A and RAD23B, MCM7 (which is involved in DNA replication), annexin A1, the PML tumor suppresso
CytoplasmNucleus
Angelman syndrome
A neurodevelopmental disorder characterized by severe motor and intellectual retardation, ataxia, frequent jerky limb movements and flapping of the arms and hands, hypotonia, seizures, absence of speech, frequent smiling and episodes of paroxysmal laughter, open-mouthed expression revealing the tongue.
Contributes to a melanosome-specific anion (chloride) current that modulates melanosomal pH for optimal tyrosinase activity required for melanogenesis and the melanosome maturation (PubMed:11310796, PubMed:15262401, PubMed:22234890, PubMed:25513726). One of the components of the mammalian pigmentary system (PubMed:15262401, PubMed:18252222, PubMed:7601462). May serve as a key control point at which ethnic skin color variation is determined. Major determinant of brown and/or blue eye color (PubMe
Melanosome membrane
Albinism, oculocutaneous, 2
An autosomal recessive disorder in which the biosynthesis of melanin pigment is reduced in skin, hair, and eyes. Although affected infants may appear at birth to have complete absence of melanin pigment, most patients acquire small amounts of pigment with age. Visual anomalies include decreased acuity and nystagmus. The phenotype is highly variable. The hair of affected individuals may turn darker with age, and pigmented nevi or freckles may be seen. African and African American individuals may have yellow hair and blue-gray or hazel irides. One phenotypic variant, 'brown OCA,' has been described in African and African American populations and is characterized by light brown hair and skin color and gray to tan irides.
Catalytic component of P4-ATPase flippase complex, which catalyzes the hydrolysis of ATP coupled to the transport of phosphatidylcholine (PC) from the outer to the inner leaflet of the plasma membrane (PubMed:25947375, PubMed:29599178, PubMed:30530492). Initiates inward plasma membrane bending and recruitment of Bin/amphiphysin/Rvs (BAR) domain-containing proteins involved in membrane tubulation and cell trafficking (PubMed:29599178). Facilitates ITGB1/beta1 integrin endocytosis, delaying cell a
Cell membraneEndoplasmic reticulum membrane
May be involved in tissue-specific alternative RNA processing events
Nucleus
Medicamentos e terapias
Mecanismo: Gamma-aminobutyric acid receptor subunit alpha-4/beta3/delta agonist
Mecanismo: Dopamine D3 receptor agonist
Mecanismo: DOPA decarboxylase inhibitor
Mecanismo: DOPA decarboxylase inhibitor
Variantes genéticas (ClinVar)
1.856 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1.753 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
6 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Angelman
Centros de Referência SUS
24 centros habilitados pelo SUS para Síndrome Angelman
Centros para Síndrome Angelman
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
17 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
52 ensaios clínicos encontrados, 18 ativos.
Publicações mais relevantes
Mostrando amostra de 200 publicações de um total de 886
Emerging disease-modifying therapies for Angelman syndrome: A comprehensive review for pediatric neurologists.
Angelman syndrome (AS), a rare neurogenetic disorder affecting approximately 1 in 15,000 live births, results from loss of functional UBE3A gene expression and manifests with severe developmental delay, intellectual disability, absent speech, ataxia, epilepsy, and distinctive behavioral features. Until recently, only symptomatic management was available. This review provides pediatric neurologists with a comprehensive, practice-oriented overview of emerging disease-modifying therapies for AS, focusing on therapeutic approaches advancing through clinical development. The molecular pathophysiology of AS, natural history considerations critical for trial interpretation, and the current evidence for antisense oligonucleotide (ASO) therapies (ION582, GTX-102/apazunersen, rugonersen), gene replacement approaches (MVX-220), and next-generation strategies including CRISPR-based gene editing, artificial transcription factors, small molecules, and novel delivery platforms are reviewed. ASO therapies targeting the UBE3A antisense transcript represent the most clinically advanced approach, with three candidates showing proof-of-concept efficacy in Phase 1/2 studies and two advancing to pivotal Phase 3 trials. Gene replacement therapy offers potential single-administration treatment but faces challenges regarding safety, immune responses, and durability. Next-generation approaches including CRISPR activation, epigenetic editing, and blood-brain barrier-penetrating delivery systems show preclinical promise. Critical challenges include outcome measurement limitations, genotype stratification, long-term safety monitoring, and ensuring equitable access. These advances herald a transformation in AS clinical care and represent a milestone in precision pediatric neurology.
Tuberous sclerosis complex.
Tuberous sclerosis complex (TSC) is a rare genetic disease caused by heterozygous loss-of-function variants in TSC1 or TSC2. Patients present with benign tumours known as hamartomas in the brain, eyes, lungs, kidneys, heart and skin. Many hamartomas contain mosaic second hit variants in TSC1 or TSC2. The most disabling features of TSC include epilepsy and TSC-associated neuropsychiatric disorders (TAND) such as intellectual disability and autism spectrum disorder. Remarkable progress has been made both in understanding the pathogenesis of TSC and in its clinical management, largely due to the discovery of the link between TSC1 and TSC2 and the mechanistic target of rapamycin (mTOR) signalling pathway. TSC1 and TSC2 form a protein complex that inhibits mTOR. Naturally occurring inhibitors of mTOR (rapamycin) and its analogues, collectively known as rapalogues, have been used to test various hypotheses in preclinical models and are approved for the treatment of several manifestations of TSC. Approved drug treatments (rapalogues) exist for subependymal giant cell astrocytomas, renal angiomyolipomas, pulmonary lymphangioleiomyomatosis, facial angiofibromas and refractory seizures. However, there is still an unmet need for effective treatment of TAND and refractory epilepsy, despite the available medical and surgical options.
Unlocking the potential of multidisciplinary clinics to transform rare epilepsies care, insights, and research.
Multidisciplinary clinics (MDCs) improve care for patients with complex, comorbid conditions through coordinated, team-based care. Despite their potential, MDCs remain underutilized and understudied in pediatric neurology, particularly for individuals with rare, chronic epilepsies. The subject of MDCs in pediatric epilepsy was explored through two workshops and surveys of caregivers and clinicians. MDC models vary widely-from general clinics (e.g., neurology, genetics, and neuropsychology) to disorder-specific clinics with multisystemic specialists. Caregivers identified key barriers, including geographical distance, personal expense, and insurance prior authorization requirements, yet overall reported positive experiences-citing valuable opportunities to participate in research and meaningful changes to clinical care. Although the findings reflect responses from a predominantly white, higher-income, English-speaking group of caregivers recruited through patient advocacy networks-and may therefore carry certain biases-their perspectives remain broadly generalizable to prospective patients across diverse socioeconomic settings. Similarly, physicians identified funding and space as the primary barriers to establishing multidisciplinary clinics, yet a majority recognized the importance of advancing research, translational studies, and clinical trials. MDCs can improve care for patients with medically complex rare epilepsies by integrating the management of comorbidities. These clinics bring value to both rare patients and physicians by providing a setting for synergistic activities between clinical care, clinical trials, and research. To expand their impact, we recommend: (1) establishing more MDCs using sustainable models; (2) improving access to extend the reach of MDCs; (3) including key specialists for integrated care; (4) sharing disorder-specific expertise through collaboration and training; and (5) tracking standardized success measures to validate and scale these efforts.
Pharmacodynamics, Efficacy, and Safety of Intraputaminal Eladocagene Exuparvovec Administered to Pediatric Patients With Aromatic L-Amino Acid Decarboxylase Deficiency Using an MR-Compatible Cannula: 48 Weeks of Follow-Up.
Aromatic ʟ-amino acid decarboxylase (AADC) deficiency is a rare pediatric neurotransmitter disorder that typically necessitates lifelong care, and that carries a risk of childhood mortality. Eladocagene exuparvovec gene therapy is designed to restore AADC production. Study GT-002 (NCT04903288) is a phase 2, multicenter, open-label trial assessing the pharmacodynamics, safety, and efficacy of eladocagene exuparvovec administered to the putamen bilaterally in pediatric patients with AADC deficiency using a magnetic resonance (MR)-compatible cannula. Patients received eladocagene exuparvovec at 1.8 × 1011 vector genomes via the SmartFlow MR-compatible cannula in a single operative session. Endpoints include the change from baseline in cerebrospinal fluid homovanillic acid levels, motor milestone achievement, and safety. Here we report results from 48 weeks of follow-up. Mean (SD) cerebrospinal fluid homovanillic acid levels increased from baseline (22.5 [32.3] nmol/L; n = 13) to week 48 (55.3 [45.6] nmol/L; change from baseline: 28.3 [13.7] nmol/L; p = 0.0003; n = 9), indicating de novo dopamine production. At baseline (n = 13), all patients showed severe motor developmental delay; at week 48 (n = 12), nine achieved full head control, four could sit unassisted, two could stand with support, and two could walk independently to a toy. Overall, 260 treatment-emergent adverse events were reported in 13 patients; 259 were deemed unrelated and one likely unrelated to the MR-compatible cannula. No treatment-emergent adverse events led to study withdrawal and no deaths occurred. This study provides further evidence of the favorable pharmacodynamic, efficacy, and safety profile of eladocagene exuparvovec in children with AADC deficiency; intraputaminal administration using an MR-compatible cannula was well tolerated. Study GT-002 (NCT04903288) provides further evidence of the favourable pharmacodynamic, efficacy and safety profile of eladocagene exuparvovec gene therapy in children with AADC deficiency over 48 weeks and demonstrates that intraputaminal administration using an MR-compatible cannula was well tolerated, allowing for real-time MRI confirmation of cannula placement and infusate coverage, and for accurate dosing to the putamen.
The serotonin receptor 7 as an emerging target to restore altered neuroplasticity in Angelman syndrome.
The serotonin receptor 7 (5-HT7R) has been indicated as a key modulator of neuronal structure and function, playing critical roles in synaptic plasticity, dendritic spine formation, and cytoskeletal remodeling. 5-HT7R activation promotes neurite outgrowth, enhances long-term potentiation (LTP), stimulates local protein synthesis at synapses, and regulates mitochondrial functions, and the mTOR pathway. These properties make the 5-HT7R a compelling candidate for therapeutic intervention in neurodevelopmental disorders characterized by synaptic dysfunctions. Angelman syndrome (AS) is a severe neurodevelopmental disorder caused by the loss of function of the maternal UBE3A gene, resulting in impairments of synaptic plasticity, dendritic spine density, protein synthesis, mitochondrial activity and mTOR signaling. Intriguingly, many of the processes altered in AS are the ones that are positively regulated by 5-HT7R activation. For instance, AS animal models exhibit reduced LTP and altered dendritic morphology and 5-HT7R stimulation enhances synaptic strength and spine formation in the brain of wild type rodents. Moreover, BDNF/TrkB function signaling is impaired and mitochondrial integrity is disrupted in AS and 5-HT7R agonists enhance the altered BDNF/TrkB signalling and restore mitochondrial dysfunctions in Rett syndrome (RTT) mice model. Interestingly, recent evidence demonstrates that pharmacological activation of 5-HT7Rs increases synaptic protein synthesis, restores LTP, enhances dendritic spine density, and improves cognitive function in an AS mouse model. These encouraging results open the way to future studies using neurons and brain organoids generated from iPSCs obtained from AS patients, which represent novel tools in preclinical research. Overall, 5-HT7R stimulation, by counteracting the molecular alterations associated with the loss of UBE3A, may represent a novel approach to restore neural function in the mature brain, leading to translational applications in AS patients, and possibly also in other synaptopathies. Clinical trial number: not applicable.
Publicações recentes
Real-world effectiveness of highly purified cannabidiol in epilepsy associated with 15q11.2-q13.1 duplication and deletion syndromes: A multicenter study.
Association between a diagnostic journey in Angelman syndrome and caregivers' quality of life.
Proteasome dysfunction underlies HERC2-linked neurodevelopmental disorder with Angelman-like clinical features.
Ophthalmic phenotype and strabismus surgery in Angelman syndrome: genotype-specific risks and uniform surgical efficacy.
Operant behavior is reliably impaired in a mouse model of Angelman syndrome.
📚 EuropePMC1.104 artigos no totalmostrando 192
Emerging disease-modifying therapies for Angelman syndrome: A comprehensive review for pediatric neurologists.
Brain & developmentSocial support and maternal caregiving burden in families of children with Angelman syndrome in China: the mediating role of self-stigma.
BMC psychologyTuberous sclerosis complex.
Nature reviews. Disease primersA de Novo Genome Assembly of an Angelman Syndrome Pig (sus Scrofa Domesticus) Model to Resolve SNHG14.
The Journal of heredityCrossing the finish line towards a disease-modifying treatment for Angelman syndrome.
Journal of neurodevelopmental disordersUnlocking the potential of multidisciplinary clinics to transform rare epilepsies care, insights, and research.
Frontiers in neurologyRNAi-Induced Expression of Paternal UBE3A.
GenesCaregiver Meaningful Score Differences and Meaningful Score Regions for the Observer-Reported Communication Ability Measure for Individuals with Angelman Syndrome.
Value in health : the journal of the International Society for Pharmacoeconomics and Outcomes ResearchPharmacodynamics, Efficacy, and Safety of Intraputaminal Eladocagene Exuparvovec Administered to Pediatric Patients With Aromatic L-Amino Acid Decarboxylase Deficiency Using an MR-Compatible Cannula: 48 Weeks of Follow-Up.
Journal of inherited metabolic diseaseDevelopment of at-home video recordings for functional skill assessment in Angelman Syndrome: a pilot study.
Journal of neurodevelopmental disordersCommunicative Development Inventories (CDIs) in etiologically diverse developmental conditions: A systematic review.
Research in developmental disabilitiesThe serotonin receptor 7 as an emerging target to restore altered neuroplasticity in Angelman syndrome.
Experimental neurologyClinical Presentation, Genetics, and Laboratory Testing with Integrated Genetic Analysis of Molecular Mechanisms in Prader-Willi and Angelman Syndromes: A Review.
International journal of molecular sciencesAdvances and challenges of precision epigenetic therapy in treating genomic imprinting diseases.
Translational pediatricsUBE3A isoform-selective and non-selective contributions to Angelman syndrome phenotypes.
Molecular psychiatryA dual-reporter mouse for therapeutic discovery in Angelman syndrome.
JCI insightAdjunctive cannabidiol in intractable pediatric epilepsy: A retrospective study on tolerability, efficacy, and safety across genetic and nongenetic etiologies.
MedicineAssociation of the Observer-Reported Communication Ability (ORCA) Measure with Established Communication Measures: Insights From the Angelman Syndrome Natural History Study.
Journal of autism and developmental disordersA systematic review of highly purified cannabidiol in developmental and epileptic encephalopathies and complex treatment-resistant epilepsies: Changes in seizure frequency and adverse events.
Epilepsy researchPredictors of communication ability in Angelman syndrome: evidence from Polish individuals.
Journal of communication disordersLoss of Drosophila UBE3A phenocopies Piezo dysfunction and drives hyperphagic feeding in Drosophila.
FlyThe Italian Angelman Syndrome Registry (IReAS): a tool for standardized data collection and genotype-phenotype analysis.
European journal of medical geneticsDeveloping Meaningful Score Differences for the Bayley-4 and Vineland-3 in Angelman Syndrome Using a Delphi Panel.
Value in health : the journal of the International Society for Pharmacoeconomics and Outcomes ResearchGeneration of human pineal gland organoids with melatonin production for disease modeling.
Cell stem cellMeasuring economic burden in families of individuals with Angelman Syndrome in Poland: a caregivers' survey.
Orphanet journal of rare diseasesClinical and Genetic Profiles of 11 Chinese Patients With Angelman Syndrome.
Genetics researchMicrostructural White Matter Alterations in Angelman Syndrome: A Fixel-Based Analysis.
Autism research : official journal of the International Society for Autism ResearchExtracellular vesicle dysfunction contributes to synaptic and cognitive deficits in a mouse model of Angelman syndrome.
Progress in neurobiologyAdvancing observer-reported outcome measurement: development of the MOOD-AS for observing distress in Angelman syndrome.
Journal of patient-reported outcomesLong-Term Modulation of Cortical Excitability by Repeated Anodal Transcranial Direct Current Stimulation Highlights Neurobiological Constraints in a Neurodevelopmental Disorder Model.
Neuromodulation : journal of the International Neuromodulation SocietyPsychological and psychiatric service use among family caregivers of individuals with Angelman Syndrome: A cross-sectional study.
Psychiatria polskaUBE3A stabilization of β-catenin preserves synaptic proteins essential for motor and cognitive functions in Angelman Syndrome.
Molecular autismDysfunction of the Autophagy System and MDM2-p53 Axis Leads to the Accumulation of Amyloidogenic Proteins in Angelman Syndrome Models.
International journal of molecular sciencesBrain-wide Genome Editing via STEP-RNPs for Treatment of Angelman Syndrome.
bioRxiv : the preprint server for biologyAd astra per aspera: treatment challenges and opportunities for children with spinal muscular atrophy and tracheostomy.
Gene therapyMulti-targeting zinc finger nuclease vector unsilences paternal UBE3A in a mouse model of Angelman syndrome.
Gene therapyBehavioral and molecular insights into anxiety in ube3a and fmr1 zebrafish models of autism spectrum disorders.
Translational psychiatryRNA-based therapies for neurodevelopmental disorders: innovative tools for molecular correction.
Frontiers in molecular biosciencesAngelman Syndrome With Papillary Urothelial Carcinoma: An Unusual Initial Presentation and Two Recurrences.
CureusDevelopment of the Angelman syndrome video assessment: quantifying meaningful change.
Journal of neurodevelopmental disordersA prodrug targeting CIM6P/IGF2R enhances memory in healthy mice and reverses deficits in an Angelman syndrome mouse model.
Translational psychiatryExperience-Dependent Plasticity to Visual Sequences in Mouse Anterior Cingulate Cortex Reflects Familiarity.
The Journal of neuroscience : the official journal of the Society for NeuroscienceImprinting Disorders and Epigenetic Alterations in Children Conceived by Assisted Reproductive Technologies: Mechanisms, Clinical Outcomes, and Prenatal Diagnosis.
GenesA luminescence-based biosensor to measure endogenous UBE3A activity.
iScienceEfficacy and tolerability of a low-glycemic-index ketogenic diet in Angelman syndrome: findings from the DIANE study.
Orphanet journal of rare diseasesNIPA1 depletion in tumor-associated macrophages via IGFBP2/EGFR attenuates acute myeloid leukemia progression and chemoresistance.
Annals of hematologyCriterion Validity, Scalability and Stability of Scoring on the Bayley-III in Children With Angelman Syndrome.
Journal of intellectual disability research : JIDRCofilin Inhibition Ameliorates PIEZO2 and AMPA Dysfunction in a Mouse Model of Angelman Syndrome.
The Journal of neuroscience : the official journal of the Society for NeuroscienceCourtship and distress ultrasonic vocalizations are altered in a mouse model of Angelman syndrome.
Journal of neurodevelopmental disordersPeriodic and aperiodic contributions to EEG delta power are translatable and complementary Angelman syndrome biomarkers.
bioRxiv : the preprint server for biologyOpen-label phase IV trial evaluating nusinersen after onasemnogene abeparvovec in children with spinal muscular atrophy.
The Journal of clinical investigationA stem cell-based toolkit to model Angelman syndrome caused by paternal uniparental disomy of chromosome 15.
Human cellDysregulation of Neuronal Activity-Dependent Immediate Early Genes in a Mouse Model of Angelman Syndrome.
Journal of neurochemistryA human Angelman Syndrome class II pluripotent stem cell line with fluorescent paternal UBE3A reporter.
Frontiers in cell and developmental biologyBehavior Decoding Delineates Seizure Microfeatures and Associated Sudden Death Risks in Mouse Models of Epilepsy.
Annals of neurologyCell Type-Specific Contributions of UBE3A to Angelman Syndrome Behavioral Phenotypes.
eNeuroMutation of ube3a causes developmental abnormalities and autism-like molecular and behavioral alterations in zebrafish.
Brain research bulletinAAV-dCas9 vector unsilences paternal Ube3a in neurons by impeding Ube3a-ATS transcription.
Communications biologyMeasuring Childhood Disability Using the National Health Interview Survey.
JAMA pediatricsParental Stress and Family Quality of Life in Families of Individuals Living With Angelman Syndrome.
Journal of intellectual disability research : JIDREnvironmental context modulates sociability in ube3a zebrafish mutants via alterations in sensory pathways.
Molecular psychiatryAngelman syndrome patient-derived neuron screen leads to clinical ASO rugonersen targeting UBE3A-ATS with long-lasting effect in monkeys.
Nucleic acids researchAtypical alpha oscillatory EEG dynamics in children with Angelman syndrome.
NeuroImage. ClinicalUBE3A reinstatement restores behaviorand proteome in an Angelman syndrome mouse model of imprinting defects.
Molecular autismIdentification of CNKSR2 Pathogenic Variant and Detection of Strong XCI in a Female Patient With Severe DEE-SWAS and Phenotype Expansion in Male Patients.
Clinical geneticsIncreased Extra-Axial Cerebrospinal Fluid Volume in Children with Angelman Syndrome: Links to Sleep Problems and Seizures.
Annals of the Child Neurology SocietyAssessment of Dysphagia in Chinese Cohort of Angelman Syndrome: An Observational Study.
CNS neuroscience & therapeuticsA pig model of Angelman syndrome.
Lab animalEarly-Onset 15q11.2 Microdeletion Syndrome in a Six-Year-Old Child: A Case Report of Refractory Epilepsy, Autism, and Multisystem Manifestations.
CureusHigh-throughput assessment of FMR1 and SNRPN methylation-based newborn screening using IsoPure and QIAcube HT systems.
EpigenomicsProlonged Penile Erection in an Adolescent with Angelman Syndrome under Aripiperazole: A Case Report.
Iranian journal of child neurologyOutcomes After Tonsillectomy in Children With Angelman Syndrome.
Journal of otolaryngology - head & neck surgery = Le Journal d'oto-rhino-laryngologie et de chirurgie cervico-facialePaternal UPD (15) With Disease-Causing Mutation and Small Supernumerary Ring Chromosome 15: A Case Report.
Case reports in geneticsPreclinical pharmacology of alogabat: a novel GABAA-α5 positive allosteric modulator targeting neurodevelopmental disorders with impaired GABAA signaling.
Frontiers in pharmacology[Example of a genetic condition caused by an imprinting disorder: Angelman syndrome].
Annales de biologie cliniqueDrug Repurposing Patent Applications October-December 2024.
Assay and drug development technologiesA preclinical pig model of Angelman syndrome mirrors the early developmental trajectory of the human condition.
Proceedings of the National Academy of Sciences of the United States of AmericaConformational flexibility and rotational binding shifts in W104C mutant MECP2-DNA complexes: A molecular simulation study.
Biochemical and biophysical research communicationsCannabidiol Intervention for Non-Epileptic Myoclonus in Angelman Syndrome: A Case Report.
Movement disorders clinical practiceThe Ubiquitin E3 Ligase UBE3A Regulates GRIPAP1 and PACSIN1 Proteins Linked to the Endocytic Recycling of AMPA Receptors.
Molecular and cellular biologyPrelinguistic Communication Complexity of Children With Neurogenetic Syndromes.
Journal of speech, language, and hearing research : JSLHRThe UBE3A-ATS antisense oligonucleotide rugonersen in children with Angelman syndrome: a phase 1 trial.
Nature medicineAngelman Syndrome: Multidisciplinary Management.
Journal of pediatric health care : official publication of National Association of Pediatric Nurse Associates & PractitionersMechanism of EHMT2-mediated genomic imprinting associated with Prader-Willi syndrome.
Nature communicationsAssociation Between Feeding Problems and Gastrointestinal Symptoms, Language, and Developmental History in Adults With Angelman Syndrome.
American journal of medical genetics. Part AOral health care of people with Angelman syndrome in Germany - a questionnaire-based study.
BMC oral healthSignificance of Behavioural Problems on Family Impact in Angelman Syndrome: A Prospective Cross-Sectional Study.
Journal of autism and developmental disordersLanguage comprehension assessment using the computer-based instrument for low motor language testing (C-BiLLT) in children with Angelman syndrome.
Augmentative and alternative communication (Baltimore, Md. : 1985)Diagnosis of Angelman Syndrome, With 66 Years of Delay, Using Hypothesis-Free DNA Methylation Profiling.
Clinical geneticsEpileptic seizures and EEG findings in 3p deletion syndrome involving SLC6A1.
European journal of medical geneticsEmerging roles for E3 ubiquitin ligases in neural development and disease.
Frontiers in cell and developmental biologyHealth outcomes of children with Prader-Willi or Angelman syndromes: a European population-based multicentre study.
Archives of disease in childhoodWhen Vomiting Isn't Just a Bug: Unmasking Two Rare Causes of Pediatric Dysphagia.
CureusUsing the Bayley-4 and Vineland-3 in Angelman syndrome: barriers, solutions, and challenging items.
Orphanet journal of rare diseasesCaregiving Burden and Quality of Life Among Parents of Individuals With Angelman Syndrome: Gender Differences and the Impact of Financial Well-Being.
Pediatric neurologyLoss of UBE3A impacts both neuronal and non-neuronal cells in human cerebral organoids.
Communications biologyHexasomy of the 15q11q13 region: a detailed report and review of the literature.
European journal of medical geneticsGeneration of two pairs of induced pluripotent stem cells from Angelman syndrome patients with class I 15q11.2-q13.1 deletions and their unaffected familial controls.
Stem cell researchRemote EEG acquisition in Angelman syndrome using PANDABox-EEG.
Journal of neurodevelopmental disordersNovel method for detection of UBE3A protein in CSF from individuals with Angelman syndrome.
Molecular genetics and metabolismFacial expression deep learning algorithms in the detection of neurological disorders: a systematic review and meta-analysis.
Biomedical engineering onlineAcute administration of lovastatin had no pronounced effect on motor abilities, motor coordination, gait nor simple cognition in a mouse model of Angelman syndrome.
Journal of neurodevelopmental disorders[Sleep disorders in imprinting disorders].
Zhurnal nevrologii i psikhiatrii imeni S.S. KorsakovaMyoclonic Dystonia: A Common Phenomenology in the Pleomorphic Movements of Angelman Syndrome.
Movement disorders clinical practiceAutism and intellectual disability due to a novel gain-of-function mutation in UBE3A.
Journal of human geneticsSmall Supernumerary Marker Chromosome (sSMC) 15 in Male Primary Infertility: A Case Study.
Case reports in medicineActivity-dependent degradation of Kv4.2 contributes to synaptic plasticity and behavior in Angelman syndrome model mice.
Cell reportsClinical Efficacy and Safety of the Ketogenic Diet in Patients with Genetic Confirmation of Drug-Resistant Epilepsy.
NutrientsDietary Intake of Octanoic Acid Restores UBE3A Expression and Improves the Behavioral Phenotypes in a Mouse Model of Angelman Syndrome.
FASEB journal : official publication of the Federation of American Societies for Experimental BiologyDynamic shift in localization of UBE3A across developmental stages in an Angelman syndrome mouse model.
Neurobiology of diseaseMom genes and dad genes: genomic imprinting in the regulation of social behaviors.
EpigenomicsNew Insights into Chromosomal Regions 15p11.2-15q11.2 by Studying Submicroscopic Variations Using Molecular Cytogenetics.
Cytogenetic and genome researchLocalization of human UBE3A isoform 3 is highly sensitive to amino acid substitutions at p.Met21 position.
Human molecular geneticsLower respiratory rate during sleep in children with angelman syndrome compared to age-matched controls.
Orphanet journal of rare diseasesEpilepsy associated with chromosomal disorders.
Epilepsy & behavior : E&BMolecular aspects of Angelman Syndrome: Defining the new path forward.
Biomolecules & biomedicineActivity-dependent regulation of Cdc42 by Ephexin5 drives synapse growth and stabilization.
Science advancesAge-Related Trajectories of Autistic Traits in Children With Angelman Syndrome.
Autism research : official journal of the International Society for Autism ResearchThe gain-of-function UBE3AQ588E variant causes Angelman-like neurodevelopmental phenotypes in mice.
Scientific reportsUnraveling the Roles of UBE3A in Neurodevelopment and Neurodegeneration.
International journal of molecular sciencesForestwalk: A Machine Learning Workflow Brings New Insights Into Posture and Balance in Rodent Beam Walking.
The European journal of neuroscienceGelastic spells in Angelman Syndrome, when laughter isn't funny.
Epilepsy & behavior reportsCourtship and distress ultrasonic vocalizations are disrupted in a mouse model of Angelman syndrome.
Research squareThe economic impact of caregiving for individuals with Angelman syndrome in the United States: results from a caregiver survey.
Orphanet journal of rare diseasesPerception of psychosocial burden in mothers of children with rare pediatric neurological diseases.
Scientific reportsParents' Experiences and Views About Use of Wearable Technology for Research and Treatment Monitoring of Children with Neurodevelopmental Disorders.
Journal of developmental and behavioral pediatrics : JDBPA high sensitivity assay of UBE3A ubiquitin ligase activity.
Methods (San Diego, Calif.)Perception of four intellectual and developmental disabilities based on search engine and news portrayal.
PloS oneBinDel: Detecting Clinically Relevant Fetal Genomic Microdeletions Using Low-Coverage Whole-Genome Sequencing-Based NIPT.
Prenatal diagnosisUBE3A controls axon initial segment in the cortical pyramidal neurons.
Biochemical and biophysical research communicationsWearable sensors in paediatric neurology.
Developmental medicine and child neurologyBranched endosomal disruptor (BEND) lipids mediate delivery of mRNA and CRISPR-Cas9 ribonucleoprotein complex for hepatic gene editing and T cell engineering.
Nature communicationsGenetics of Prader-Willi and Angelman syndromes: 2024 update.
Current opinion in psychiatryProteomic Profiling of Potential E6AP Substrates via Ubiquitin-based Photo-Crosslinking Assisted Affinity Enrichment.
Chembiochem : a European journal of chemical biologyThree-Dimensional Gait Analysis of School-Age Children With Angelman Syndrome: A Case-Control Study.
American journal of medical genetics. Part AClinical and Cytogenetic Impact of Maternal Balanced Double Translocation: A Familial Case of 15q11.2 Microduplication and Microdeletion Syndromes with Genetic Counselling Implications.
GenesConcordance of Whole-Genome Long-Read Sequencing with Standard Clinical Testing for Prader-Willi and Angelman Syndromes.
The Journal of molecular diagnostics : JMDDifferences in structure, dynamics, and zinc coordination between isoforms of human ubiquitin ligase UBE3A.
The Journal of biological chemistryHyperkaliaemic cardiac arrest in Angelman's syndrome following suxamethonium.
BMJ case reportsComparative profiling of white matter development in the human and mouse brain reveals volumetric deficits and delayed myelination in Angelman syndrome.
Molecular autism[Autosomal dominant intellectual developmental disorder 60 with seizures: a case report].
Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatricsHeterozygous UBR5 variants result in a neurodevelopmental syndrome with developmental delay, autism, and intellectual disability.
American journal of human geneticsUniparental disomy (UPD) exclusion in embryos following Preimplantation Genetic Testing for Structural Rearrangements (PGT-SR).
Journal of assisted reproduction and geneticsCommunity-Sourced Reporting of Mortalities in Angelman Syndrome (1979-2022).
American journal of medical genetics. Part ACaMKIIα hub ligands are unable to reverse known phenotypes in Angelman syndrome mice.
Basic & clinical pharmacology & toxicologySurgical Treatment of Strabismus in Children With Developmental Delay: A Review of the Literature and Results of Personal Experience.
Journal of pediatric ophthalmology and strabismusThe Breastfeeding Experiences of Mother-Infant Dyads and the Effects of an FMR1 Mutation.
Journal of autism and developmental disordersUse of Basket Trials to Solve Sleep Problems in Patients with Rare Diseases.
Clocks & sleepOptimization of the activity and biodegradability of ionizable lipids for mRNA delivery via directed chemical evolution.
Nature biomedical engineeringLong-read sequencing for detection and subtyping of Prader-Willi and Angelman syndromes.
Journal of medical geneticsPeptidomimetic inhibitors targeting TrkB/PSD-95 signaling improves cognition and seizure outcomes in an Angelman Syndrome mouse model.
Neuropsychopharmacology : official publication of the American College of NeuropsychopharmacologyUBE3A: Bridging the gap between neurodevelopment, neural function, and neurodegenerative woes.
Journal of Alzheimer's disease : JADGeneration of isogenic models of Angelman syndrome and Prader-Willi syndrome in CRISPR/Cas9-engineered human embryonic stem cells.
PloS oneGenome-wide screening reveals essential roles for HOX genes and imprinted genes during caudal neurogenesis of human embryonic stem cells.
Stem cell reportsAlterations of synaptic plasticity in Angelman syndrome model mice are rescued by 5-HT7R stimulation.
Progress in neurobiologyWidespread Gene Editing in the Brain via In Utero Delivery of mRNA Using Acid-Degradable Lipid Nanoparticles.
ACS nanoParents' perspectives on conversations about prognosis and an assessment of prognostic information available online: A mixed-methods study.
Disability and health journal[Epilepsy in Angelman syndrome and the most common electroencephalographic findings].
Revista de neurologiaAutism Spectrum Disorder Symptom Profiles in Fragile X Syndrome, Angelman Syndrome, Tuberous Sclerosis Complex and Neurofibromatosis Type 1.
Journal of autism and developmental disordersComprehensive molecular and clinical findings in 29 patients with multi-locus imprinting disturbance.
Clinical epigeneticsAAV vector-derived elements integrate into Cas9-generated double-strand breaks and disrupt gene transcription.
Molecular therapy : the journal of the American Society of Gene TherapyEpigenomic newborn screening for conditions with intellectual disability and autistic features in Australian newborns.
EpigenomicsAgenesis of the Ductus Venosus and Its Association With Genetic Abnormalities.
Prenatal diagnosisCombinatorial design of siloxane-incorporated lipid nanoparticles augments intracellular processing for tissue-specific mRNA therapeutic delivery.
Nature nanotechnology[Angelman syndrome: current approach and the future of therapies].
MedicinaMultiscale spatio-temporal dynamics of UBE3A gene in brain physiology and neurodevelopmental disorders.
Neurobiology of diseaseEpigenetic editing alleviates Angelman syndrome phenotype in mice by unsilencing paternal Ube3a.
Cell discoveryValidation of high-sensitivity assays to quantitate cerebrospinal fluid and serum β-galactosidase activity in patients with GM1-gangliosidosis.
Molecular therapy. Methods & clinical developmentAssociation between sleep disturbances and challenging behavior in children and adolescents with Angelman syndrome.
Sleep medicineDeciphering the physiopathology of neurodevelopmental disorders using brain organoids.
Brain : a journal of neurologyEvaluation of an In-House Genetic Testing Method for Confirming Prader-Willi and Angelman Syndromes in Sri Lanka.
Clinical laboratorySuccessful use of cannabidiol in nonconvulsive status epilepticus in Angelman syndrome.
Epilepsia openAngelman syndrome in Poland: current diagnosis and therapy status-the caregiver perspective: a questionnaire study.
Orphanet journal of rare diseasesGenotype-phenotype correlation over time in Angelman syndrome: Researching 134 patients.
HGG advancesPrecision Medicine in Angelman Syndrome.
NeuropediatricsNeuronal UBE3A substrates hold therapeutic potential for Angelman syndrome.
Current opinion in neurobiologyMandibular Distraction in Dual Syndromic Diagnosis.
The Journal of craniofacial surgeryEpigenetics in rare neurological diseases.
Frontiers in cell and developmental biologyA case of an Angelman-syndrome caused by an intragenic duplication of UBE3A uncovered by adaptive nanopore sequencing.
Clinical epigeneticsDo metabolic deficits contribute to sleep disruption in monogenic intellectual disability syndromes?
Trends in neurosciences1H-NMR-based metabolomics reveals metabolic alterations in early development of a mouse model of Angelman syndrome.
Molecular autismIntegration of CTCF loops, methylome, and transcriptome in differentiating LUHMES as a model for imprinting dynamics of the 15q11-q13 locus in human neurons.
Human molecular geneticsA clinical-translational review of sleep problems in neurodevelopmental disabilities.
Journal of neurodevelopmental disordersFast and facile synthesis of amidine-incorporated degradable lipids for versatile mRNA delivery in vivo.
Nature chemistryUbe3a unsilencer for the potential treatment of Angelman syndrome.
Nature communicationsExploring the Clinical and Genetic Landscape of Angelman Syndrome: Patient-Reported Insights from an Italian Registry.
Journal of clinical medicinePopulation-Based Risk of Psychiatric Disorders Associated With Recurrent Copy Number Variants.
JAMA psychiatryAge-Related Blood Levels of Creatine Kinase-MM in Newborns and Patients with Duchenne Muscular Dystrophy: Considerations for the Development of Newborn Screening Algorithms.
International journal of neonatal screeningInterlinked destinies: How ubiquitin-proteasome and autophagy systems underpin neurocognitive outcomes.
Experimental neurologyDevelopmental milestones and daily living skills in individuals with Angelman syndrome.
Journal of neurodevelopmental disordersRegional and cellular organization of the autism-associated protein UBE3A/E6AP and its antisense transcript in the brain of the developing rhesus monkey.
Frontiers in neuroanatomyGeneration of three iPSC lines with inducible systems to be used in Angelman syndrome research.
Stem cell researchAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Angelman.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Angelman
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Emerging disease-modifying therapies for Angelman syndrome: A comprehensive review for pediatric neurologists.
- Tuberous sclerosis complex.
- Unlocking the potential of multidisciplinary clinics to transform rare epilepsies care, insights, and research.
- Pharmacodynamics, Efficacy, and Safety of Intraputaminal Eladocagene Exuparvovec Administered to Pediatric Patients With Aromatic L-Amino Acid Decarboxylase Deficiency Using an MR-Compatible Cannula: 48 Weeks of Follow-Up.
- The serotonin receptor 7 as an emerging target to restore altered neuroplasticity in Angelman syndrome.
- Real-world effectiveness of highly purified cannabidiol in epilepsy associated with 15q11.2-q13.1 duplication and deletion syndromes: A multicenter study.
- Association between a diagnostic journey in Angelman syndrome and caregivers' quality of life.
- Proteasome dysfunction underlies HERC2-linked neurodevelopmental disorder with Angelman-like clinical features.
- Ophthalmic phenotype and strabismus surgery in Angelman syndrome: genotype-specific risks and uniform surgical efficacy.
- Operant behavior is reliably impaired in a mouse model of Angelman syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:72(Orphanet)
- OMIM OMIM:105830(OMIM)
- MONDO:0007113(MONDO)
- GARD:5810(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q535364(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
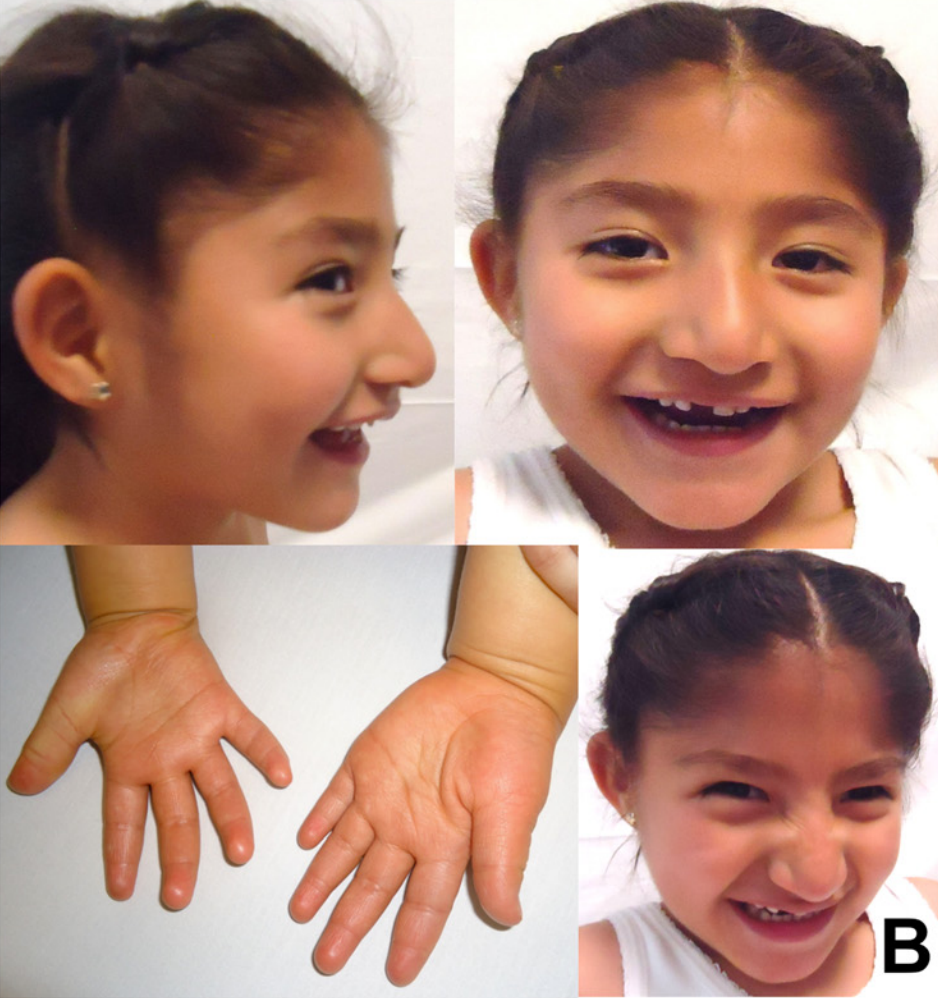
Síndrome Angelman
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos (literatura)
- fonte: Orphanet
- Ensaios clínicos
- fonte: ClinicalTrials.gov