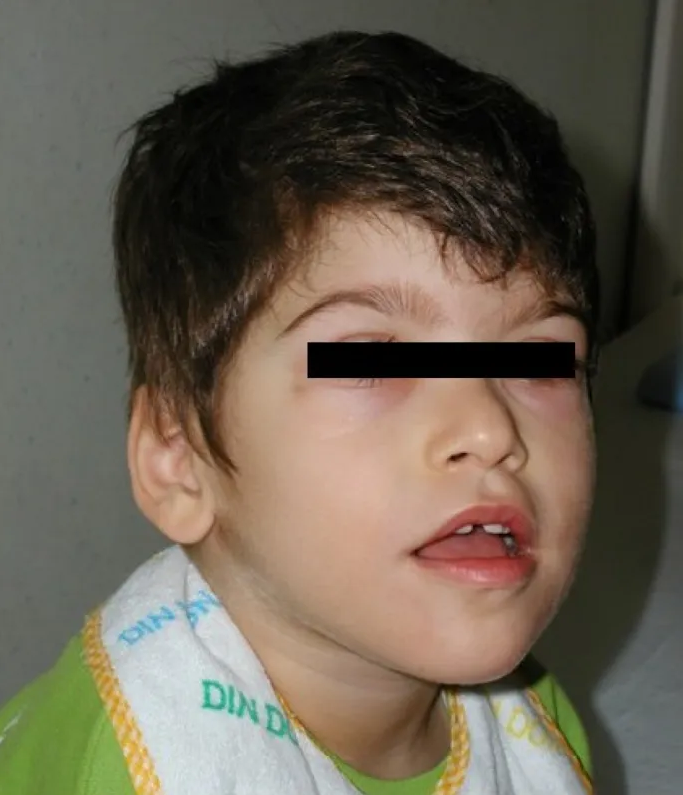
Síndrome rara autossômica recessiva associada ao gene PIGG, caracterizada por epilepsia de início precoce, retardo do desenvolvimento intelectual severo, atraso no crescimento intrauterino e anomalias cerebrais como hipoplasia cerebelar. Apresenta também desequilíbrio da marcha, fala ausente ou atrasada, e características faciais e esqueléticas específicas.
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome de epilepsia de início precoce-transtorno do desenvolvimento intelectual-anomalias cerebrais é uma doença genética rara que afeta o desenvolvimento neurológico. Ela se caracteriza por convulsões que começam nos primeiros dias de vida (período neonatal), atraso global do desenvolvimento intelectual e alterações na estrutura do cérebro, como a hipoplasia (desenvolvimento incompleto) do cerebelo e do corpo caloso. A condição é tão rara que sua prevalência estimada é de menos de 1 caso para cada 1.000.000 de pessoas.[1][4]
Sinais e sintomas
Os sinais e sintomas da síndrome são variados e afetam múltiplos sistemas. Os principais incluem: convulsões (crises tônico-clônicas bilaterais), atraso ou ausência da fala, dificuldades para andar (ataxia da marcha e desequilíbrio), hipotonia (tônus muscular baixo), hipermobilidade articular (especialmente nos dedos dos pés e das mãos), e características faciais como ponte nasal deprimida e lábio superior fino. Também podem ocorrer comportamentos do espectro autista, espasmos musculares, hipermetropia (dificuldade para enxergar de perto), dificuldades alimentares e retardo do crescimento intrauterino.[1][4]
Causas genéticas
A síndrome é causada por mutações no gene PIGG (sigla em inglês para 'GPI ethanolamine phosphate transferase 2, catalytic subunit'). Esse gene fornece instruções para a produção de uma enzima essencial para a síntese de uma âncora de glicosilfosfatidilinositol (GPI), uma estrutura que fixa diversas proteínas na superfície das células. Quando o gene PIGG não funciona corretamente, esse processo é prejudicado, afetando o desenvolvimento e a função do sistema nervoso. A herança é autossômica recessiva, o que significa que a criança precisa herdar uma cópia do gene alterado de cada um dos pais para desenvolver a doença.[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sintomas e confirmado por testes genéticos. O sequenciamento completo do exoma (WES) é o principal exame para identificar mutações no gene PIGG. Além disso, exames de imagem do cérebro (como ressonância magnética) podem mostrar achados como hipoplasia cerebelar, atrofia cerebelar e hipoplasia do corpo caloso. O eletroencefalograma (EEG) frequentemente revela padrões anormais, como espículas focais. Exames laboratoriais, como a dosagem de aminoácidos e ácidos orgânicos na urina, ajudam a descartar outros erros inatos do metabolismo. O teste do pezinho (triagem neonatal) pode detectar alguns distúrbios metabólicos, mas não é específico para esta síndrome. Atualmente, há 335 variantes genéticas associadas à doença registradas no ClinVar, um banco de dados público de variantes.[1][2][5]
Tratamento e manejo
Não existe cura para a síndrome, e o tratamento é multidisciplinar e focado no manejo dos sintomas e na melhora da qualidade de vida. O controle das convulsões é feito com medicamentos antiepilépticos, que devem ser prescritos e ajustados por um neurologista. O atraso no desenvolvimento e as dificuldades motoras e de fala são abordados com fisioterapia, terapia ocupacional e fonoaudiologia. O acompanhamento com oftalmologista é importante para corrigir a hipermetropia. O suporte nutricional pode ser necessário para lidar com as dificuldades alimentares. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura completa para o atendimento, incluindo procedimentos como a dosagem de aminoácidos, o sequenciamento do exoma e o atendimento em reabilitação para doenças raras.[1][2]
Tratamentos citados na literatura
A literatura científica menciona diversos fármacos que foram estudados ou utilizados em contextos relacionados a esta síndrome, mas é importante destacar que estas são associações mineradas de publicações científicas e não representam uma recomendação de tratamento. Os fármacos listados incluem: aminohydroxybutyric-acid, diclofenamide, ethosuximide, felbamate, fosphenytoin, lamotrigine, metharbital, phenacemide, pyridoxal, stiripentol, topiramate, valproic-acid, valpromide e zonisamide. A decisão sobre o uso de qualquer medicamento deve ser tomada exclusivamente pelo médico assistente, com base na avaliação individual de cada paciente.[1][2]
Prognóstico e qualidade de vida
O prognóstico varia de acordo com a gravidade dos sintomas e a resposta ao tratamento. Como a doença se manifesta desde o nascimento, o acompanhamento precoce e contínuo com uma equipe multidisciplinar é fundamental para maximizar o potencial de desenvolvimento e minimizar complicações. O suporte familiar e o acesso a serviços de reabilitação e educação especializada são pilares essenciais para promover a melhor qualidade de vida possível para a criança e sua família.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Síndrome rara autossômica recessiva associada ao gene PIGG, caracterizada por epilepsia de início precoce, retardo do desenvolvimento intelectual severo, atraso no crescimento intrauterino e anomalias cerebrais como hipoplasia cerebelar. Apresenta também desequilíbrio da marcha, fala ausente ou atrasada, e características faciais e esqueléticas específicas.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome de epilepsia de início precoce-transtorno do desenvolvimento intelectual-anomalias cerebrais é uma doença genética rara que afeta o desenvolvimento neurológico. Ela se caracteriza por convulsões que começam nos primeiros dias de vida (período neonatal), atraso global do desenvolvimento intelectual e alterações na estrutura do cérebro, como a hipoplasia (desenvolvimento incompleto) do cerebelo e do corpo caloso. A condição é tão rara que sua prevalência estimada é de menos de 1 caso para cada 1.000.000 de pessoas.[1][4]
Sinais e sintomas
Os sinais e sintomas da síndrome são variados e afetam múltiplos sistemas. Os principais incluem: convulsões (crises tônico-clônicas bilaterais), atraso ou ausência da fala, dificuldades para andar (ataxia da marcha e desequilíbrio), hipotonia (tônus muscular baixo), hipermobilidade articular (especialmente nos dedos dos pés e das mãos), e características faciais como ponte nasal deprimida e lábio superior fino. Também podem ocorrer comportamentos do espectro autista, espasmos musculares, hipermetropia (dificuldade para enxergar de perto), dificuldades alimentares e retardo do crescimento intrauterino.[1][4]
Causas genéticas
A síndrome é causada por mutações no gene PIGG (sigla em inglês para 'GPI ethanolamine phosphate transferase 2, catalytic subunit'). Esse gene fornece instruções para a produção de uma enzima essencial para a síntese de uma âncora de glicosilfosfatidilinositol (GPI), uma estrutura que fixa diversas proteínas na superfície das células. Quando o gene PIGG não funciona corretamente, esse processo é prejudicado, afetando o desenvolvimento e a função do sistema nervoso. A herança é autossômica recessiva, o que significa que a criança precisa herdar uma cópia do gene alterado de cada um dos pais para desenvolver a doença.[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sintomas e confirmado por testes genéticos. O sequenciamento completo do exoma (WES) é o principal exame para identificar mutações no gene PIGG. Além disso, exames de imagem do cérebro (como ressonância magnética) podem mostrar achados como hipoplasia cerebelar, atrofia cerebelar e hipoplasia do corpo caloso. O eletroencefalograma (EEG) frequentemente revela padrões anormais, como espículas focais. Exames laboratoriais, como a dosagem de aminoácidos e ácidos orgânicos na urina, ajudam a descartar outros erros inatos do metabolismo. O teste do pezinho (triagem neonatal) pode detectar alguns distúrbios metabólicos, mas não é específico para esta síndrome. Atualmente, há 335 variantes genéticas associadas à doença registradas no ClinVar, um banco de dados público de variantes.[1][2][5]
Tratamento e manejo
Não existe cura para a síndrome, e o tratamento é multidisciplinar e focado no manejo dos sintomas e na melhora da qualidade de vida. O controle das convulsões é feito com medicamentos antiepilépticos, que devem ser prescritos e ajustados por um neurologista. O atraso no desenvolvimento e as dificuldades motoras e de fala são abordados com fisioterapia, terapia ocupacional e fonoaudiologia. O acompanhamento com oftalmologista é importante para corrigir a hipermetropia. O suporte nutricional pode ser necessário para lidar com as dificuldades alimentares. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura completa para o atendimento, incluindo procedimentos como a dosagem de aminoácidos, o sequenciamento do exoma e o atendimento em reabilitação para doenças raras.[1][2]
Tratamentos citados na literatura
A literatura científica menciona diversos fármacos que foram estudados ou utilizados em contextos relacionados a esta síndrome, mas é importante destacar que estas são associações mineradas de publicações científicas e não representam uma recomendação de tratamento. Os fármacos listados incluem: aminohydroxybutyric-acid, diclofenamide, ethosuximide, felbamate, fosphenytoin, lamotrigine, metharbital, phenacemide, pyridoxal, stiripentol, topiramate, valproic-acid, valpromide e zonisamide. A decisão sobre o uso de qualquer medicamento deve ser tomada exclusivamente pelo médico assistente, com base na avaliação individual de cada paciente.[1][2]
Prognóstico e qualidade de vida
O prognóstico varia de acordo com a gravidade dos sintomas e a resposta ao tratamento. Como a doença se manifesta desde o nascimento, o acompanhamento precoce e contínuo com uma equipe multidisciplinar é fundamental para maximizar o potencial de desenvolvimento e minimizar complicações. O suporte familiar e o acesso a serviços de reabilitação e educação especializada são pilares essenciais para promover a melhor qualidade de vida possível para a criança e sua família.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 16 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 43 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisCatalytic subunit of the ethanolamine phosphate transferase 2 complex that transfers an ethanolamine phosphate (EtNP) from a phosphatidylethanolamine (PE) to the 6-OH position of the second alpha-1,6-linked mannose of a 2-acyl-6-[6-phosphoethanolamine-alpha-D-mannosyl-(1->2)-alpha-D-mannosyl-(1->6)-2-phosphoethanolamine-alpha-D-mannosyl-(1->4)-alpha-D-glucosaminyl]-1-(1-radyl,2-acyl-sn-glycero-3-phospho)-1D-myo-inositol (also termed H7) intermediate to generate a 2-acyl-6-[6-phosphoethanolamine-
Endoplasmic reticulum membrane
Neurodevelopmental disorder with or without hypotonia, seizures, and cerebellar atrophy
An autosomal recessive disorder characterized by delayed psychomotor development, hypotonia, and early-onset seizures in most patients. Additional variable features are cerebellar atrophy, ataxia, and non-specific dysmorphic features. Some patients may have the Emm-null blood group phenotype.
Variantes genéticas (ClinVar)
335 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de epilepsia de início precoce-transtorno do desenvolvimento intelectual-anomalias cerebrais
Centros de Referência SUS
37 centros habilitados pelo SUS para Síndrome de epilepsia de início precoce-transtorno do desenvolvimento intelectual-anomalias cerebrais
Centros para Síndrome de epilepsia de início precoce-transtorno do desenvolvimento intelectual-anomalias cerebrais
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
2 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Publicações recentes
Mast cell mediators in hereditary angioedema.
Prenatal Molecular Diagnosis of COL2A1-Associated Stickler Syndrome: Genotype-Phenotype Correlation in a Resource-Limited Healthcare Setting.
Platelet gene signatures detecting pulmonary artery stenosis in patients with pulmonary hypertension.
The global impact of imiglucerase therapy in children with Gaucher disease types 1 and 3: a real-world analysis from the International Collaborative Gaucher Group Gaucher Registry.
Monogenic lupus with SLC7A7 mutations: a retrospective study from a Chinese center.
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de epilepsia de início precoce-transtorno do desenvolvimento intelectual-anomalias cerebrais.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de epilepsia de início precoce-transtorno do desenvolvimento intelectual-anomalias cerebrais
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Mast cell mediators in hereditary angioedema.
- Prenatal Molecular Diagnosis of COL2A1-Associated Stickler Syndrome: Genotype-Phenotype Correlation in a Resource-Limited Healthcare Setting.
- Platelet gene signatures detecting pulmonary artery stenosis in patients with pulmonary hypertension.
- The global impact of imiglucerase therapy in children with Gaucher disease types 1 and 3: a real-world analysis from the International Collaborative Gaucher Group Gaucher Registry.
- Monogenic lupus with SLC7A7 mutations: a retrospective study from a Chinese center.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:488635(Orphanet)
- OMIM OMIM:616917(OMIM)
- MONDO:0014832(MONDO)
- Epilepsia(PCDT · Ministério da Saúde)
- GARD:17897(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q56014612(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de epilepsia de início precoce-transtorno do desenvolvimento intelectual-anomalias cerebrais
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub