Doença peroxissomal caracterizada por desmielinização episódica progressiva, polineuropatia sensório-motora e perda auditiva que tem base material em mutação heterozigótica no gene ACOX1 no cromossomo 17q25.1.
Introdução
O que você precisa saber de cara
Doença peroxissomal caracterizada por desmielinização episódica progressiva, polineuropatia sensório-motora e perda auditiva que tem base material em mutação heterozigótica no gene ACOX1 no cromossomo 17q25.1.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 5 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 14 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisInvolved in the initial and rate-limiting step of peroxisomal beta-oxidation of straight-chain saturated and unsaturated very-long-chain fatty acids (PubMed:15060085, PubMed:17458872, PubMed:17603022, PubMed:32169171, PubMed:33234382, PubMed:7876265). Catalyzes the desaturation of fatty acyl-CoAs that have a saturated bond between C2 and C3 (2,3-saturated acyl-CoA) to 2-trans-enoyl-CoAs ((2E)-enoyl-CoAs), and donates electrons directly to molecular oxygen (O(2)), thereby producing hydrogen perox
Peroxisome
Adrenoleukodystrophy, pseudoneonatal
A peroxisomal single-enzyme disorder of fatty acid beta-oxidation, resulting in clinical manifestations that remind neonatal adrenoleukodystrophy. Clinical features include intellectual disability, leukodystrophy, seizures, mild hepatomegaly, hearing deficit. Pseudo-NALD is characterized by increased plasma levels of very-long chain fatty acids, due to decreased or absent peroxisome acyl-CoA oxidase activity. Peroxisomes are intact and functioning.
Variantes genéticas (ClinVar)
125 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 42 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
5 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Mitchell
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
[A clinical case of erythromelalgia in a patient comorbid with antiphospholipid syndrome].
The article presents a clinical case of secondary erythromelalgia in a comorbid patient with antiphospholipid syndrome, accompanied by burning pain, hyperemia and swelling of the lower extremities. A comprehensive examination involving a neurologist, dermatologist, and rheumatologist was conducted to establish the diagnosis and select the optimal therapy. Significant clinical improvement in the patient's condition is demonstrated against the background of pregabalin therapy. The importance of a multidisciplinary approach and the difficulty of diagnosis due to the lack of specific examination methods and insufficient awareness of doctors about this disease are emphasized. The described case demonstrates the need for a personalized approach to managing patients with erythromelalgia to improve their quality of life. В статье представлен клинический случай вторичной эритромелалгии у коморбидной пациентки с антифосфолипидным синдромом, сопровождающейся жгучей болью, гиперемией и отеком нижних конечностей. Представлено комплексное обследование пациентки с привлечением невролога, дерматолога и ревматолога для установки диагноза и подбора оптимальной терапии. Продемонстрировано значимое клиническое улучшение состояния пациентки на фоне терапии прегабалином. Подчеркивается важность мультидисциплинарного подхода и трудность диагностики, обусловленные отсутствием специфических методов обследования и недостаточной осведомленностью врачей о таком заболевании. Описанный случай демонстрирует необходимость персонализированного подхода к ведению пациентов с эритромелалгией для улучшения качества их жизни.
Dermatopathological features and successful treatment with topical antioxidant for ichthyosiform lesions in Mitchell syndrome caused by an ACOX1 variant.
Peroxisomal acyl-CoA oxidase 1 (ACOX1), is a peroxisomal enzyme that catalyzes β-oxidation of very-long-chain fatty acids (VLCFA). The gain-of-function variant p.Asn237Ser in ACOX1 has been shown to cause Mitchell syndrome (MITCH), a neurodegenerative disorder characterized by episodic demyelination, hearing loss, and polyneuropathy, through the overproduction of hydrogen peroxide. Only eight cases of MITCH have been reported. While all these patients experienced cutaneous abnormalities, detailed skin features and potential treatment have not been documented. Herein, we report two MITCH patients who harbored a de novo heterozygous variant p.Asn237Ser in ACOX1 and experienced progressive ichthyosiform erythroderma. Skin histopathology revealed hyperkeratosis and parakeratosis with focal hypogranulosis as well as dyskeratotic keratinocytes. Lipid accumulation in the epidermis was observed using Oil Red O staining. Both patients exhibited a remarkable response to treatment with the topical antioxidant N-acetylcysteine (NAC), with Patient 1 achieving complete recovery after 3 months of consistent treatment. This study provides the first comprehensive description of the clinicopathological characteristics and effective treatment of skin lesions in MITCH patients. The successful treatment with topical NAC suggests excessive reactive oxygen species might play a significant role in the pathogenesis of skin lesions in MITCH.
Exploiting fly models to investigate rare human neurological disorders.
Rare neurological diseases, while individually are rare, collectively impact millions globally, leading to diverse and often severe neurological symptoms. Often attributed to genetic mutations that disrupt protein function or structure, understanding their genetic basis is crucial for accurate diagnosis and targeted therapies. To investigate the underlying pathogenesis of these conditions, researchers often use non-mammalian model organisms, such as Drosophila (fruit flies), which is valued for their genetic manipulability, cost-efficiency, and preservation of genes and biological functions across evolutionary time. Genetic tools available in Drosophila, including CRISPR-Cas9, offer a means to manipulate gene expression, allowing for a deep exploration of the genetic underpinnings of rare neurological diseases. Drosophila boasts a versatile genetic toolkit, rapid generation turnover, and ease of large-scale experimentation, making it an invaluable resource for identifying potential drug candidates. Researchers can expose flies carrying disease-associated mutations to various compounds, rapidly pinpointing promising therapeutic agents for further investigation in mammalian models and, ultimately, clinical trials. In this comprehensive review, we explore rare neurological diseases where fly research has significantly contributed to our understanding of their genetic basis, pathophysiology, and potential therapeutic implications. We discuss rare diseases associated with both neuron-expressed and glial-expressed genes. Specific cases include mutations in CDK19 resulting in epilepsy and developmental delay, mutations in TIAM1 leading to a neurodevelopmental disorder with seizures and language delay, and mutations in IRF2BPL causing seizures, a neurodevelopmental disorder with regression, loss of speech, and abnormal movements. And we explore mutations in EMC1 related to cerebellar atrophy, visual impairment, psychomotor retardation, and gain-of-function mutations in ACOX1 causing Mitchell syndrome. Loss-of-function mutations in ACOX1 result in ACOX1 deficiency, characterized by very-long-chain fatty acid accumulation and glial degeneration. Notably, this review highlights how modeling these diseases in Drosophila has provided valuable insights into their pathophysiology, offering a platform for the rapid identification of potential therapeutic interventions. Rare neurological diseases involve a wide range of expression systems, and sometimes common phenotypes can be found among different genes that cause abnormalities in neurons or glia. Furthermore, mutations within the same gene may result in varying functional outcomes, such as complete loss of function, partial loss of function, or gain-of-function mutations. The phenotypes observed in patients can differ significantly, underscoring the complexity of these conditions. In conclusion, Drosophila represents an indispensable and cost-effective tool for investigating rare neurological diseases. By facilitating the modeling of these conditions, Drosophila contributes to a deeper understanding of their genetic basis, pathophysiology, and potential therapies. This approach accelerates the discovery of promising drug candidates, ultimately benefiting patients affected by these complex and understudied diseases.
ACOX1 gain-of-function post-mortem neuropathology is distinct from ACOX1 loss-of-function: case report and literature review.
Generation and characterization of a zebrafish gain-of-function ACOX1 Mitchell disease model.
Mitchell syndrome is a rare, neurodegenerative disease caused by an ACOX1 gain-of-function mutation (c.710A>G; p.N237S), with fewer than 20 reported cases. Affected patients present with leukodystrophy, seizures, and hearing loss. ACOX1 serves as the rate-limiting enzyme in peroxisomal beta-oxidation of very long-chain fatty acids. The N237S substitution has been shown to stabilize the active ACOX1 dimer, resulting in dysregulated enzymatic activity, increased oxidative stress, and glial damage. Mitchell syndrome lacks a vertebrate model, limiting insights into the pathophysiology of ACOX1-driven white matter damage and neuroinflammatory insults. We report a patient presenting with rapidly progressive white matter damage and neurological decline, who was eventually diagnosed with an ACOX1 N237S mutation through whole genome sequencing. We developed a zebrafish model of Mitchell syndrome using transient ubiquitous overexpression of the human ACOX1 N237S variant tagged with GFP. We assayed zebrafish behavior, oligodendrocyte numbers, expression of white matter and inflammatory transcripts, and analysis of peroxisome counts. The patient experienced progressive leukodystrophy and died 2 years after presentation. The transgenic zebrafish showed a decreased swimming ability, which was restored with the reactive microglia-targeted antioxidant dendrimer-N-acetyl-cysteine conjugate. The mutants showed no effect on oligodendrocyte counts but did display activation of the integrated stress response (ISR). Using a novel SKL-targeted mCherry reporter, we found that mutants had reduced density of peroxisomes. We developed a vertebrate (zebrafish) model of Mitchell syndrome using transient ubiquitous overexpression of the human ACOX1 N237S variant. The transgenic mutants exhibited motor impairment and showed signs of activated ISR, but interestingly, there were no changes in oligodendrocyte counts. However, the mutants exhibited a deficiency in the number of peroxisomes, suggesting a possible shared mechanism with the Zellweger spectrum disorders.
Publicações recentes
[A clinical case of erythromelalgia in a patient comorbid with antiphospholipid syndrome].
ACOX1 gain-of-function post-mortem neuropathology is distinct from ACOX1 loss-of-function: case report and literature review.
ACOX1 gain-of-function variation in a 10-years-old patient responsive to immunomodulating therapy.
Dermatopathological features and successful treatment with topical antioxidant for ichthyosiform lesions in Mitchell syndrome caused by an ACOX1 variant.
Exploiting fly models to investigate rare human neurological disorders.
📚 EuropePMC2 artigos no totalmostrando 9
[A clinical case of erythromelalgia in a patient comorbid with antiphospholipid syndrome].
Terapevticheskii arkhivACOX1 gain-of-function post-mortem neuropathology is distinct from ACOX1 loss-of-function: case report and literature review.
Free neuropathologyACOX1 gain-of-function variation in a 10-years-old patient responsive to immunomodulating therapy.
American journal of medical genetics. Part ADermatopathological features and successful treatment with topical antioxidant for ichthyosiform lesions in Mitchell syndrome caused by an ACOX1 variant.
The Journal of dermatologyExploiting fly models to investigate rare human neurological disorders.
Neural regeneration researchGeneration and characterization of a zebrafish gain-of-function ACOX1 Mitchell disease model.
Frontiers in pediatricsACOX1 Gain-of-Function Variant in Two German Pediatric Patients, in One Case Mimicking Autoimmune Inflammatory Disease.
NeuropediatricsA de novo heterozygous variant in ACOX1 gene cause Mitchell syndrome: the first case in China and literature review.
BMC medical genomicsReal-time quaking-induced conversion assays for prions: Applying a sensitive but imperfect test in clinical practice.
European journal of neurologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Mitchell.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Mitchell
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- [A clinical case of erythromelalgia in a patient comorbid with antiphospholipid syndrome].
- Dermatopathological features and successful treatment with topical antioxidant for ichthyosiform lesions in Mitchell syndrome caused by an ACOX1 variant.
- Exploiting fly models to investigate rare human neurological disorders.
- ACOX1 gain-of-function post-mortem neuropathology is distinct from ACOX1 loss-of-function: case report and literature review.
- Generation and characterization of a zebrafish gain-of-function ACOX1 Mitchell disease model.
- ACOX1 gain-of-function variation in a 10-years-old patient responsive to immunomodulating therapy.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:631248(Orphanet)
- OMIM OMIM:618960(OMIM)
- MONDO:0030073(MONDO)
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q122917581(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
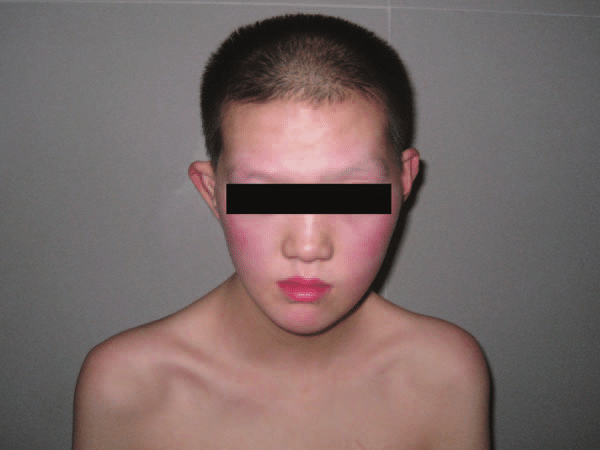
Síndrome Mitchell
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- Dado público estruturado
- fonte: Wikidata