A displasia tricodentoóssea (TDO) é um tipo de displasia ectodérmica, caracterizada por cabelo cacheado ou crespo desde o nascimento, esmalte dos dentes malformado e com descoloração, taurodontismo nos molares, ossos com maior densidade que o normal (DMO) e ossos do crânio mais espessos.
Introdução
O que você precisa saber de cara
A displasia tricodentoóssea (TDO) é um tipo de displasia ectodérmica, caracterizada por cabelo cacheado ou crespo desde o nascimento, esmalte dos dentes malformado e com descoloração, taurodontismo nos molares, ossos com maior densidade que o normal (DMO) e ossos do crânio mais espessos.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 8 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 18 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisTranscriptional activator (By similarity). Activates transcription of GNRHR, via binding to the downstream activin regulatory element (DARE) in the gene promoter (By similarity)
NucleusCytoplasm
Trichodentoosseous syndrome
An autosomal dominant disease characterized by curly kinky hair at birth, enamel hypoplasia, taurodontism, thickening of cortical bones and variable expression of craniofacial morphology.
Variantes genéticas (ClinVar)
25 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 11 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome trico-dento-ósseo
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Differential Effects of DLX3 Mutations Drive Phenotypic Variability in Tricho-Dento-Osseous Syndrome via Direct Activation of WNT10A.
DLX3 is a homeobox transcription factor essential for multiple organogenesis processes. Mutations in DLX3 cause trichodentoosseous syndrome (TDO), characterized by curly hair, sclerotic bone, enamel, and dentin defects as well as taurodontism. Phenotypic variability in TDO has been well documented, but its pathogenesis remains poorly understood. Here, we characterized three TDO families with distinct clinical features and identified a known DLX3 deletion (c.561_562del) and the first pathogenic splice-site variant (c.516+1_516+2insA). The proband with the splice-site mutation displayed a mesenchymal-dominant phenotype with severe dentin hypoplasia, enlarged pulp chambers, and hypertaurodontism but nearly normal enamel, whereas the mother and sister showed epithelial-dominant anomalies, including enamel hypoplasia and kinky hair. Minigene analysis demonstrated that c.516+1_516+2insA generated two aberrant transcripts encoding p.Val173Aspfs*28 and p.Arg120_Val173del. These mutant proteins localized mainly in the cytoplasm and showed markedly reduced transactivation activity. In cultured human dental pulp cells, DLX3 overexpression upregulated the odontoblastic markers DSPP, MMP20, and WNT10A. Chromatin immunoprecipitation and reporter assays further revealed that DLX3 directly activates WNT10A via a conserved enhancer (chr2:218,878,973_218,879,302) and three upstream binding sites. These findings expand the TDO mutational spectrum and suggest that differential mutant DLX3 expression may contribute to phenotypic variability, whereas disrupted regulation of WNT10A underlies dentin defects and taurodontism.
Dento-Craniofacial Features of Tricho-Dento-Osseous Syndrome: A Systematic Review and Meta-Analysis.
Tricho-dento-osseous syndrome (TDOS), a rare autosomal dominant condition caused by mutations in DLX3, is characterized by abnormalities in teeth, bone, and hair. This systematic review and meta-analysis summarized the most frequently reported dento-craniofacial features of TDOS. Searches were undertaken in five databases supplemented by manual scrutiny and a gray literature search. Observational and descriptive studies were included. Risk of bias was appraised using the Joanna Briggs Institute tools. Meta-analyses of continuous, binary, and proportion data were performed, with results reported as odds ratio (OR) and 95% confidence intervals (CI). Twenty-seven studies describing 297 individuals with TDOS were included. Most studies demonstrated a low risk of bias. Taurodontism (70.5%), enamel hypoplasia (34.5%), and dental infections (28.7%) were the most prevalent dental findings. Increased bone density/thickness (43.8%) was the primary skeletal manifestation, and sparse hair (27.2%) was the most common hair abnormality. Meta-analyses revealed high odds for taurodontism (OR = 42.71; 95% CI = 7.45-244.75) and consistent prevalence estimates for taurodontism (73%; 95% CI = 0.52-0.97) and enamel hypoplasia (71%; 95% CI = 0.52-0.97). Data confirm that TDOS predominantly affects dental, skeletal, and hair structures, highlighting the need for early diagnosis, multidisciplinary care, and tailored treatment approaches.
Structural Variants in COL1A1 and COL1A2 in Osteogenesis Imperfecta.
Osteogenesis Imperfecta (OI) is a heterogeneous skeletal dysplasia characterized by bone fragility, skeletal deformities, and short stature. Most commonly, it is caused by autosomal dominant variants in the type I collagen genes, COL1A1 or COL1A2. Type I collagen is the main protein of the extracellular matrix in the skeleton and changes in its structure or quantity may lead to OI. 85%-90% of OI cases occur due to sequence variants in type I collagen genes, while OI caused by structural abnormalities in type I collagen genes is less common. In most cases, haploinsufficiency of type I collagen is associated with a milder OI phenotype. Large genomic deletions often involve several genes within the same chromosomal region, leading to microdeletion syndromes with OI features. Here, we report eight Swedish patients from five unrelated families with OI due to structural variants in the COL1A1 and COL1A2 genes. One patient with OI type III had a complex rearrangement with a deletion and duplication event in COL1A2, leading to reduced COL1A2 expression. Three other patients from two different families with OI type I had whole gene deletions involving COL1A1. In one family, three affected individuals with OI type I had a small intragenic deletion of exons 11-12 in COL1A2. One patient had a 2.1 Mb de novo deletion encompassing COL1A1 and DLX3 genes and features of OI and tricho-dento-osseous syndrome. Overall, this study highlights the importance of investigating gene dosage abnormalities in patients with OI and further delineates clinical and genetic variability of OI caused by structural variants in type I collagen genes.
Wispy, dystrophic hair in a pediatric patient: Expanding the differential with tricho-dento-osseus syndrome.
Craniofacial syndromes and class III phenotype: common genotype fingerprints? A scoping review and meta-analysis.
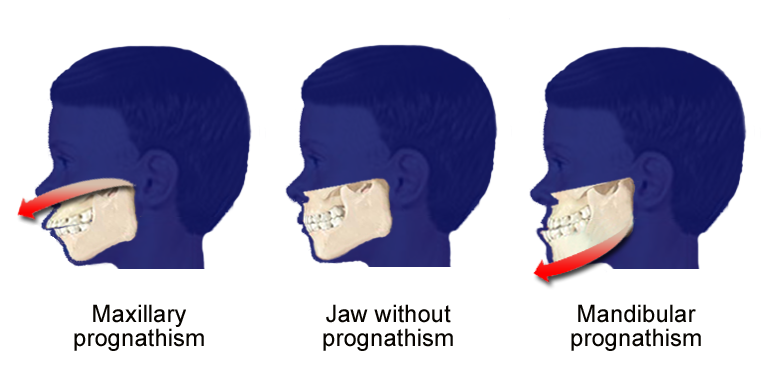
Skeletal Class III (SCIII) is among the most challenging craniofacial dysmorphologies to treat. There is, however, a knowledge gap regarding which syndromes share this clinical phenotype. The aims of this study were to: (i) identify the syndromes affected by the SCIII phenotype; (ii) clarify the involvement of maxillary and/or mandibular structures; (iii) explore shared genetic/molecular mechanisms. A two-step strategy was designed: [Step#1] OMIM, MHDD, HPO, GeneReviews and MedGen databases were explored; [Step#2]: Syndromic conditions indexed in [Step#1] were explored in Medline, Pubmed, Scopus, Cochrane Library, WOS and OpenGrey. Eligibility criteria were defined. Individual studies were assessed for risk of bias using the New Ottawa Scale. For quantitative analysis, a meta-analysis was conducted. This scoping review is a hypothesis-generating research. Twenty-two studies met the eligibility criteria. Eight syndromes affected by the SCIII were targeted: Apert syndrome, Crouzon syndrome, achondroplasia, X-linked hypohidrotic ectodermal dysplasia (XLED), tricho-dento-osseous syndrome, cleidocranial dysplasia, Klinefelter and Down syndromes. Despite heterogeneity between studies [p < 0.05], overall effects showed that midface components were affected in Apert and Down Syndromes, lower face in Klinefelter Syndrome and midface and lower face components in XLED. Our review provides new evidence on the craniofacial characteristics of genetically confirmed syndromes exhibiting the SCIII phenotype. Four major regulatory pathways might have a modulatory effect on this phenotype. IMPACT: What does this review add to the existing literature? To date, there is no literature exploring which particular syndromes exhibit mandibular prognathism as a common trait. Through this research, it was possibly to identify the particular syndromes that share the skeletal Class III phenotype (mandibular prognathism) as a common trait highlighting the common genetic and molecular pathways between different syndromes acknowledging their impact in craniofacial development.
Publicações recentes
Differential Effects of DLX3 Mutations Drive Phenotypic Variability in Tricho-Dento-Osseous Syndrome via Direct Activation of WNT10A.
Wispy, dystrophic hair in a pediatric patient: Expanding the differential with tricho-dento-osseus syndrome.
Dento-Craniofacial Features of Tricho-Dento-Osseous Syndrome: A Systematic Review and Meta-Analysis.
Structural Variants in COL1A1 and COL1A2 in Osteogenesis Imperfecta.
Craniofacial syndromes and class III phenotype: common genotype fingerprints? A scoping review and meta-analysis.
📚 EuropePMC39 artigos no totalmostrando 25
Differential Effects of DLX3 Mutations Drive Phenotypic Variability in Tricho-Dento-Osseous Syndrome via Direct Activation of WNT10A.
Annals of the New York Academy of SciencesWispy, dystrophic hair in a pediatric patient: Expanding the differential with tricho-dento-osseus syndrome.
JAAD case reportsDento-Craniofacial Features of Tricho-Dento-Osseous Syndrome: A Systematic Review and Meta-Analysis.
Special care in dentistry : official publication of the American Association of Hospital Dentists, the Academy of Dentistry for the Handicapped, and the American Society for Geriatric DentistryStructural Variants in COL1A1 and COL1A2 in Osteogenesis Imperfecta.
American journal of medical genetics. Part ACraniofacial syndromes and class III phenotype: common genotype fingerprints? A scoping review and meta-analysis.
Pediatric researchA novel DLX3 mutation causes tricho-dento-osseous syndrome with abnormal enamel structure and formation.
Archives of oral biologyNon-syndromic generalised hypotaurodontism in a case of Stage III Grade C periodontitis.
BMJ case reportsWoolly hair in tricho-dento-osseous syndrome.
Pediatric dermatologySalt Dependence of DNA Binding Activity of Human Transcription Factor Dlx3.
International journal of molecular sciencesNovel DLX3 variant identified in a family with tricho-dento-osseous syndrome.
Archives of oral biologyDental management of tricho-dento-osseous syndrome in adolescent patients: Literature review and case presentation.
Dental research journalAn unusual case of tricho-dento-osseous syndrome.
Dental research journal"Isolated" Amelogenesis Imperfecta Associated with DLX3 Mutation: A Clinical Case.
Case reports in geneticsNovel DLX3 variants in amelogenesis imperfecta with attenuated tricho-dento-osseous syndrome.
Oral diseases17q21.32-q22 Deletion in a girl with osteogenesis imperfecta, tricho-dento-osseous syndrome, and intellectual disability.
Congenital anomaliesmiR-675 promotes odontogenic differentiation of human dental pulp cells by epigenetic regulation of DLX3.
Experimental cell researchDLX3 promotes bone marrow mesenchymal stem cell proliferation through H19/miR-675 axis.
Clinical science (London, England : 1979)A de novo germline mutation of DLX3 in a Brown Swiss calf with tricho-dento-osseus-like syndrome.
Veterinary dermatologyDLX3 interacts with GCM1 and inhibits its transactivation-stimulating activity in a homeodomain-dependent manner in human trophoblast-derived cells.
Scientific reportsTricho-dento-osseous syndrome and precocious eruption.
Journal of clinical and experimental dentistryDLX3 mutation negatively regulates odontogenic differentiation of human dental pulp cells.
Archives of oral biologySenescence: novel insight into DLX3 mutations leading to enhanced bone formation in Tricho-Dento-Osseous syndrome.
Scientific reportsDLX3 negatively regulates osteoclastic differentiation through microRNA-124.
Experimental cell researchMorphological analyses and a novel de novo DLX3 mutation associated with tricho-dento-osseous syndrome in a Chinese family.
European journal of oral sciencesTranscriptional factor DLX3 promotes the gene expression of enamel matrix proteins during amelogenesis.
PloS oneAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome trico-dento-ósseo.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome trico-dento-ósseo
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Differential Effects of DLX3 Mutations Drive Phenotypic Variability in Tricho-Dento-Osseous Syndrome via Direct Activation of WNT10A.
- Dento-Craniofacial Features of Tricho-Dento-Osseous Syndrome: A Systematic Review and Meta-Analysis.Special care in dentistry : official publication of the American Association of Hospital Dentists, the Academy of Dentistry for the Handicapped, and the American Society for Geriatric Dentistry· 2025· PMID 40402097mais citado
- Structural Variants in COL1A1 and COL1A2 in Osteogenesis Imperfecta.
- Wispy, dystrophic hair in a pediatric patient: Expanding the differential with tricho-dento-osseus syndrome.
- Craniofacial syndromes and class III phenotype: common genotype fingerprints? A scoping review and meta-analysis.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:3352(Orphanet)
- OMIM OMIM:190320(OMIM)
- MONDO:0008592(MONDO)
- GARD:7799(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q7841074(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome trico-dento-ósseo
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata