Doença hereditária caracterizada pelo desenvolvimento de cistos que afetam os ductos coletores. Está frequentemente associada ao envolvimento hepático.
Introdução
O que você precisa saber de cara
Doença hereditária caracterizada pelo desenvolvimento de cistos que afetam os ductos coletores. Está frequentemente associada ao envolvimento hepático.
Tem tratamento?
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 23 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 61 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisPromotes ciliogenesis in renal epithelial cells and therefore participates in the tubules formation and/ or ensures the maintenance of the architecture of the lumen of the kidney (By similarity). Has an impact on cellular symmetry by ensuring correct bipolar cell division through the regulation of centrosome duplication and mitotic spindle assembly and by maintaining oriented cell division (OCD) during tubular elongation through planar cell polarity (PCP) pathway (PubMed:20554582). During epithe
Cell membraneCytoplasmCell projection, ciliumCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, spindleChromosome, centromereApical cell membraneNucleusSecreted, extracellular exosomeSecretedEndoplasmic reticulumGolgi apparatus
Polycystic kidney disease 4, with or without polycystic liver disease
A severe form of polycystic kidney disease affecting the kidneys and, in some cases, the hepatic biliary tract. The clinical spectrum is widely variable, with most cases presenting during infancy. The fetal phenotypic features classically include enlarged and echogenic kidneys, as well as oligohydramnios secondary to a poor urine output. Up to 50% of the affected neonates die shortly after birth, as a result of severe pulmonary hypoplasia and secondary respiratory insufficiency. In the subset that survives the perinatal period, morbidity and mortality are mainly related to severe systemic hypertension, renal insufficiency, and portal hypertension due to portal-tract fibrosis. PKD4 inheritance is autosomal recessive.
Involved in primary cilium formation (PubMed:19852954, PubMed:28530676). Probably acts as a transition zone protein required for localization of PKD1/PC1 and PKD2/PC2 to the ciliary membrane (PubMed:28530676)
Cytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriole
Polycystic kidney disease 5
A form of polycystic kidney disease, a disorder characterized by progressive formation and enlargement of cysts in both kidneys, typically leading to end-stage renal disease in adult life. Cysts may also occur in other organs, particularly the liver. PKD5 inheritance is autosomal recessive.
Medicamentos e terapias
Mecanismo: Receptor protein-tyrosine kinase erbB-2 inhibitor
Variantes genéticas (ClinVar)
1.839 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 4.811 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença renal policística autossômica recessiva
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
6 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
9 ensaios clínicos encontrados, 6 ativos.
Publicações mais relevantes
Deciphering the Impact of RAC1-SPTAN1 in ARPKD Cystogenesis Using Multifaceted Models.
Autosomal recessive polycystic kidney disease (ARPKD) leads to severe renal cysts and progressive kidney dysfunction, with no approved treatments. The absence of such cystic phenotypes in Pkhd1-/- mice underscores the need for novel models that better recapitulate the human disease. We developed kidney organoid-on-chip models that mimic patients' distal-nephron cysts, identifying RAC1/c-FOS as potential therapeutic targets. However, critical questions remain regarding RAC1 activation during cyst formation, cyst origins, and underlying molecular mechanisms. Using a multifaceted approach, organoid-on-chip models, transgenic mice, and patient kidney samples, we identified reduced levels of SPTAN1, a cytoskeletal spectrin protein, as a key regulator of RAC1 activation and cystic pathology. SPTAN1-mutant kidney organoids and mice exhibited distal-nephron cysts, and elevated RAC1/c-FOS expression, consistent with ARPKD patients. Transcriptomics and live imaging revealed altered calcium signaling and increased intracellular calcium. Single-cell RNA-seq identified SLC8A1, a sodium/calcium exchanger, as a marker distinguishing distal/connecting tubules from collecting ducts in human kidneys, predominantly expressed in cystic epithelia in organoids and human ARPKD kidneys. Restoring SPTAN1 in PKHD1-/- organoids via CRISPR activation alleviated cystic phenotypes, normalized intracellular calcium, and reduced RAC1/c-FOS expression. These findings position SPTAN1 as a central player in ARPKD pathogenesis and highlight epigenome editing as a potential therapeutic strategy.
Congenital Hepatic Fibrosis and/or Autosomal Recessive Polycystic Kidney Disease: A Single-center Experience.
Congenital hepatic fibrosis (CHF) and/or autosomal recessive polycystic kidney disease (ARPKD) represent rare and complex clinical conditions in childhood. Diagnostic challenges often arise due to heterogeneity in clinical manifestations. This study included pediatric patients diagnosed with CHF and/or ARPKD who were followed by the Pediatric Gastroenterology and Pediatric Nephrology Departments. Patient records were reviewed retrospectively. A total of 23 patients were included in the study. The median age of the cohort was 12.7±4.8 years, and the median age at diagnosis was 0.6±3.4 years. Thirteen patients had combined CHF and ARPKD, while 10 had isolated ARPKD. The diagnosis was incidental in 13 patients (56.5%), whereas five patients (21.7%) presented with an abdominal mass. Most patients had mutations in the polycystic kidney and hepatic disease 1 gene. Bilateral kidney enlargement and multiple millimetric cysts were identified in the majority of cases. Three patients required organ transplantation during follow-up. Two patients who underwent liver or kidney transplantation experienced no complications, whereas the patient who received combined liver and kidney transplantation developed kidney failure secondary to reflux nephropathy. Except for one patient who died in infancy, disease progression was generally mild in the cohort. Although kidney involvement is often predominant, hepatic complications may develop over time, particularly in patients with combined disease. A collaborative, multidisciplinary approach is essential for effectively managing the complex manifestations of these ciliopathies.
Identification of PDIA6 Mutation in a Case of Autosomal Recessive Polycystic Kidney Disease: A Case Report and Review of Literature.
Autosomal recessive polycystic kidney disease (ARPKD) is a rare but severe hereditary renal disorder characterized by bilaterally enlarged, cystic kidneys and varying degrees of hepatic fibrosis, often leading to early-onset kidney failure and significant morbidity. While most ARPKD cases are linked to mutations in the PKHD1 gene, recent advances in genomic sequencing have revealed that mutations in other genes, including PDIA6, may contribute to similar phenotypes. The PDIA6 gene encodes protein disulfide isomerase A6, which plays a critical role in protein folding within the endoplasmic reticulum (ER) and in the regulation of ER stress responses. Here, we report a rare and complex case of a full-term male neonate born to consanguineous Syrian refugee parents, who presented with a clinical constellation of features including polycystic kidney disease, severe oligohydramnios, pulmonary hypoplasia, microcephaly, rib thoracic dysplasia, and global developmental delay. Genetic analysis using whole-exome sequencing identified a homozygous two-base deletion in exon 5 of the PDIA6, resulting in a premature stop codon. Early diagnosis via genomic tools is essential for prognosis, management, and genetic counseling.
Advances in pediatric kidney diffusion tensor imaging: diagnostic and functional applications.
Diffusion tensor imaging (DTI) offers a non-invasive window into kidney microstructure by measuring directional water diffusion. In pediatric populations, where early detection of kidney dysfunction is crucial, DTI shows promise for evaluating structural integrity, diagnosing conditions, and monitoring chronic diseases such as autosomal recessive polycystic kidney disease (ARPKD). This review briefly presents the principles of renal DTI, key acquisition techniques, and important nuances in applying this modality to kidney evaluation. We provide an overview of representative post-acquisition processing pipelines for diffusion tensor generation, tractography, and quantitative analysis. We then summarize current applications of DTI in assessing kidney structure, including its use in select diseases, with focused emphasis on pediatric conditions such as ureteropelvic junction obstruction (UPJO), polycystic kidney disease, and pediatric kidney transplantation. Applications for other renal disorders are also reviewed. Finally, we outline current challenges related to standardization and highlight future research directions needed to refine methodology and further establish the clinical utility of renal DTI.
Caroli disease associated with autosomal recessive polycystic kidney disease: CT imaging features of a case report.
Caroli disease (CD) is a rare congenital hepatobiliary disorder characterized by multifocal segmental dilatation of the intrahepatic bile ducts, which may involve the entire liver or only a specific region. This rare pathology is seen in a very small proportion of the global population. Clinical manifestations vary between individuals and may overlap with other hepatobiliary disorders, making diagnosis and management challenging. Due to limited understanding and experience, early diagnosis and intervention are crucial for improving survival. In our case, Caroli disease is associated with autosomal recessive polycystic kidney disease (ARPKD), both of which are autosomal recessive disorders most commonly found in infants and children, with survival into adulthood being rare.
Publicações recentes
[Research advances in the diagnosis and treatment of Polycystic kidney disease].
Pegcetacoplan-induced remission in pediatric immune-complex membranoproliferative glomerulonephritis with comorbid autosomal recessive polycystic kidney disease: a case report.
Advances in pediatric kidney diffusion tensor imaging: diagnostic and functional applications.
Caroli disease associated with autosomal recessive polycystic kidney disease: CT imaging features of a case report.
Monogenic Etiologies of Kidney Cysts in the Pediatric Population: An Observational Cohort Study.
📚 EuropePMC406 artigos no totalmostrando 197
Advances in pediatric kidney diffusion tensor imaging: diagnostic and functional applications.
Pediatric radiologyCaroli disease associated with autosomal recessive polycystic kidney disease: CT imaging features of a case report.
Radiology case reportsStudies of mice with a large deletion of the ARPKD-associated Pkhd1 locus likely explain its GWAS association with glaucoma in humans.
bioRxiv : the preprint server for biologyNext-Generation Sequencing Defines a Molecularly Confirmed ARPKD Core Within the Broader PKHD1-Associated Disease Spectrum.
GenesDeciphering the Impact of RAC1-SPTAN1 in ARPKD Cystogenesis Using Multifaceted Models.
Advanced science (Weinheim, Baden-Wurttemberg, Germany)Novel compound heterozygous PKHD1 mutations in a Chinese ARPKD pedigree and analysis of genotype-phenotype correlations.
Frontiers in medicineDisruption of the human cystin-1 myristoyl-electrostatic switch causes polycystic kidney disease that phenocopies autosomal recessive polycystic kidney disease.
Kidney internationalInvestigation of Urinary Extracellular Vesicles as Novel and Safe Therapeutics for Autosomal Recessive Polycystic Kidney Disease (ARPKD).
Journal of biomedical materials research. Part AIdentification of Pathogenic PKHD1 Variants in Infants with Autosomal Recessive Polycystic Kidney Disease from the Dhofar Region, Oman.
F1000ResearchFibrocystin/polyductin (FPC): new functional insights into ARPKD pathogenesis revealed by informatics, comparative genomics, and model systems.
Pediatric nephrology (Berlin, Germany)Congenital Hepatic Fibrosis and/or Autosomal Recessive Polycystic Kidney Disease: A Single-center Experience.
Pediatric gastroenterology, hepatology & nutritionPrenatal Diagnosis and Postnatal Outcomes of Fetal ADPKD: A Single-Center Retrospective Cohort Study.
Medicina (Kaunas, Lithuania)Clinical characteristics and genotype-phenotype correlation for patients with autosomal recessive polycystic kidney disease and PKHD1 mutations.
Clinical nephrologyPredictors of kidney survival in children with autosomal recessive polycystic kidney disease.
Clinical and experimental nephrologyOpen resection of gastroduodenal artery aneurysm with fistulization into the duodenum 17 years after coil embolization in a patient with Caroli disease.
Journal of vascular surgery cases and innovative techniquesAtypical Liver Ultrasound Image in a Boy with Autosomal Recessive Polycystic Kidney Disease (ARPKD) and New PKD1 Variant-A Case Report.
GenesCase Report: An atypical case of ARPKD highlights the utility and challenges of implementing genetic testing in cystic kidney disease.
Frontiers in pediatricsProlonged hypotension in children with bilateral kidney absence: a case series and pathophysiologic insights.
Pediatric nephrology (Berlin, Germany)Qualitative Analysis and Comparison of Externally Led Patient-Focused Drug Development Concepts for Autosomal Recessive Polycystic Kidney Disease Against SONG Initiatives.
Kidney medicineA Novel Founder PKHD1 Disease Causing Variant in Israeli Bedouins With Autosomal Recessive Polycystic Kidney Disease.
American journal of kidney diseases : the official journal of the National Kidney FoundationPrenatal diagnosis and molecular characterization of PKHD1 variants in two Chinese fetuses with Caroli disease/syndrome.
Frontiers in geneticsDeterminants of left ventricular mass in children with autosomal recessive polycystic kidney disease.
Journal of nephrologyIdentification of PDIA6 Mutation in a Case of Autosomal Recessive Polycystic Kidney Disease: A Case Report and Review of Literature.
Clinical geneticsHealth-related quality of life, mental health and caregiver burden in children with autosomal recessive polycystic kidney disease.
Pediatric nephrology (Berlin, Germany)Long-Term Peritoneal Dialysis Using a Tenckhoff Catheter in a Premature Infant With Homozygous Autosomal Recessive Polycystic Kidney Disease: A Case Report.
CureusIntronic and Coding Genetic Variants in Autosomal Recessive Polycystic Kidney Disease Among Israeli Bedouins of Arabian Peninsula Ancestry.
American journal of kidney diseases : the official journal of the National Kidney FoundationA novel gene therapy for ARPKD based on CFTR.
American journal of physiology. Gastrointestinal and liver physiologyPeritoneal Dialysis Catheters: Review of Neonatal Patient Outcomes.
The Journal of surgical researchEstimating Lifetime Risk of Autosomal Recessive Kidney Diseases Using Population-Based Genotypic Data.
Kidney international reportsKidney organoids demonstrate that PTH1R drives a cystogenic cAMP-pPKA-pCREB axis in developmental polycystic kidney disease.
American journal of physiology. Renal physiologyCardiac connections: Effects on the heart as a result of autosomal recessive polycystic kidney disease.
The Journal of physiologyCholangitis resembling Caroli's syndrome in a patient with autosomal dominant polycystic kidney disease: Case report.
SAGE open medical case reportsA Rare Diagnosis of Caroli Syndrome in a Young Patient.
Clinical case reportsUrinary peptide signature distinguishes autosomal recessive polycystic kidney disease from other causes of chronic kidney disease.
Clinical kidney journalPilot study using a discrete mathematical approach for topological analysis and ssGSEA of gene expression in autosomal recessive polycystic kidney disease.
Scientific reportsBi-allelic UGGT1 variants cause a congenital disorder of glycosylation.
American journal of human geneticsProgress, challenges, and pragmatic concessions in predicting relative risk of kidney survival in ARPKD.
Kidney internationalThe spectrum of diseases, genetic landscape and new mutation sites of hereditary cystic kidney disease.
Clinical kidney journalHeart dysfunction in a rat model with autosomal recessive polycystic kidney disease.
The Journal of physiologyRare Combination of Phenotypes of Karyomegalic Interstitial Nephritis and Autosomal Recessive Polycystic Kidney Disease in an Omani Child.
Oman medical journalThe Role of Angiotensin-II Infusion in an Infant With Autosomal Recessive Polycystic Kidney Disease Postbilateral Nephrectomies and Refractory Hypotension in the Neonatal Period.
Case reports in nephrologyPolycystic Kidney Disease in Children: The Current Status and the Next Horizon.
American journal of kidney diseases : the official journal of the National Kidney FoundationTherapeutic opportunities in polycystic kidney and liver disease through extracellular matrix dynamics.
Biochemical pharmacologyElectrolyte and metabolite composition of cystic fluid from a rat model of ARPKD.
Communications biologyA risk score to predict kidney survival in patients with autosomal recessive polycystic kidney disease at the age of two months.
Kidney internationalElucidating the Molecular Landscape of Cystic Kidney Disease: Old Friends, New Friends and Some Surprises.
American journal of medical genetics. Part AAn extracellular vesicle based hypothesis for the genesis of the polycystic kidney diseases.
Extracellular vesicleHepatopulmonary syndrome from liver disease associated with autosomal recessive polycystic kidney disease.
Pediatric nephrology (Berlin, Germany)Reduction of elevated Gli3 does not alter the progression of autosomal recessive polycystic kidney disease.
Physiological reportsUrinary Dickkopf-3 Reflects Disease Severity and Predicts Short-Term Kidney Function Decline in Renal Ciliopathies.
Kidney international reportsGene therapy in polycystic kidney disease: A promising future.
Journal of translational internal medicineMultimodal Magnetic Resonance Imaging Assessments of Kidney Disease Severity in Autosomal Recessive Polycystic Kidney Disease.
Kidney international reportsProstatic cyst in autosomal recessive polycystic kidney disease: A case presentation and literature review.
Urology case reportsShort Bowel Syndrome Is Not a Contraindication for Kidney Transplantation.
Pediatric transplantationMinigene-based splice assays provide new insights on intronic variants of the PKHD1 gene.
Human genomicsDistribution and classifications of PKHD1 gene variants in a Turkish population using the next generation sequencing method.
Turkish journal of medical sciencesPhenotypic Discordance among Siblings with Autosomal Recessive Polycystic Kidney Disease: Case Report and Review of the Literature.
NephronRefining the genetic diagnostic puzzle: A case report on a Chinese ARPKD patient with a reciprocal balanced translocation and c.2507 T > C (p.V836A) in PKHD1.
Nephrology (Carlton, Vic.)A rare cause of echogenic kidneys with oligohydramnios in the fetus: report of two different cases.
BMC pregnancy and childbirthAn infant case of autosomal recessive polycystic kidney disease-associated dilated cardiomyopathy-like hypertensive cardiomyopathy diagnosed because of urinary tract infection.
Cardiology in the young[Autosomal recessive polycystic kidney disease in a girl].
Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatricsPrimary Cilia Elongation in Early-Onset Polycystic Kidney Disease with 2 Hypomorphic PKD1 Alleles: A Case Report.
Kidney medicineNovel splice site and nonsense variants in PKHD1 cause autosomal recessive polycystic kidney disease in a Chinese Zhuang ethnic family.
MedicineGenetic landscape and clinical outcomes of autosomal recessive polycystic kidney disease in Kuwait.
HeliyonPathogenic relationship between phenotypes of ARPKD and novel compound heterozygous mutations of PKHD1.
Frontiers in geneticsA case report of autosomal recessive polycystic kidney disease with noncompaction of ventricular myocardium: coincidence or different manifestations of ciliopathy?
BMC nephrologyNext generation sequencing identifies WNT signalling as a significant pathway in Autosomal Recessive Polycystic Kidney Disease (ARPKD) manifestation and may be linked to disease severity.
Biochimica et biophysica acta. Molecular basis of diseaseVariable Clinical Presentations and Renal Outcome in Neonates With Autosomal Recessive Polycystic Kidney Disease.
CureusProteogenomics in Nephrology: A New Frontier in Nephrological Research.
Current issues in molecular biologyProtocol for the nationwide registry of patients with polycystic kidney disease: japanese national registry of PKD (JRP).
Clinical and experimental nephrologySingle-Center Experience of Pediatric Cystic Kidney Disease and Literature Review.
Children (Basel, Switzerland)Clinical manifestation, epidemiology, genetic basis, potential molecular targets, and current treatment of polycystic liver disease.
Orphanet journal of rare diseasesThe ARPKD Protein DZIP1L Regulates Ciliary Protein Entry by Modulating the Architecture and Function of Ciliary Transition Fibers.
Advanced science (Weinheim, Baden-Wurttemberg, Germany)Defects of renal tubular homeostasis and cystogenesis in the Pkhd1 knockout.
iScienceCaroli's Disease Associated with Autosomal Dominant Polycystic Kidney Disease with Acute Pancreatitis: A Case Report.
Middle East journal of digestive diseasesSurvival of Infants With Severe Congenital Kidney Disease After ECMO and Kidney Support Therapy.
PediatricsShort-Term Outcome of Isolated Kidney Transplantation in Children with Autosomal Recessive Polycystic Kidney Disease: A Case Series and Literature Review.
Clinics and practiceEpidemiology and outcomes of pediatric autosomal recessive polycystic kidney disease in the Middle East and North Africa.
Pediatric nephrology (Berlin, Germany)The Pathophysiology of Inherited Renal Cystic Diseases.
GenesA Deep Intronic PKHD1 Variant Identified by SpliceAI in a Deceased Neonate With Autosomal Recessive Polycystic Kidney Disease.
American journal of kidney diseases : the official journal of the National Kidney FoundationDifferential regulation of MYC expression by PKHD1/Pkhd1 in human and mouse kidneys: phenotypic implications for recessive polycystic kidney disease.
Frontiers in cell and developmental biologyCaroli disease with subcutaneous hemorrhage as the sole clinical manifestation: A case report.
MedicineAutosomal Recessive Polycystic Kidney Disease: Diagnosis, Prognosis, and Management.
Advances in kidney disease and healthGenetic Spectrum of Polycystic Kidney and Liver Diseases and the Resulting Phenotypes.
Advances in kidney disease and healthCombined liver-kidney transplantation in pediatric patients.
Pediatric transplantationAssessing the potential of DZIP1L gene in autosomal recessive polycystic kidney disease gene therapy.
Pediatric discoveryThe genetic spectrum of polycystic kidney disease in children.
Revista da Associacao Medica Brasileira (1992)The molecular structure and function of fibrocystin, the key gene product implicated in autosomal recessive polycystic kidney disease (ARPKD).
Annals of human geneticsComplications and prognosis of patients diagnosed with autosomal recessive polycystic kidney disease in neonatal period.
CEN case reportsFibrocystin/Polyductin releases a C-terminal fragment that translocates into mitochondria and suppresses cystogenesis.
Nature communicationsCase report: Severe hypertension-induced priapism in an infant with unrecognized autosomal recessive polycystic kidney disease.
Frontiers in pediatricsMetformin does not slow cyst growth in the PCK rat model of polycystic kidney disease.
Physiological reports[Research progress on the pathogenesis autosomal recessive polycystic kidney disease].
Zhonghua er ke za zhi = Chinese journal of pediatricsPkhd1cyli/cyli mice have altered renal Pkhd1 mRNA processing and hormonally sensitive liver disease.
Journal of molecular medicine (Berlin, Germany)Vascular Dysfunction in Polycystic Kidney Disease: A Mini-Review.
Journal of vascular researchA novel PKHD1 splicing variant identified in a fetus with autosomal recessive polycystic kidney disease.
Frontiers in geneticsClinical Characteristics and Courses of Patients With Autosomal Recessive Polycystic Kidney Disease-Mimicking Phenocopies.
Kidney international reportsCystin is required for maintaining fibrocystin (FPC) levels and safeguarding proteome integrity in mouse renal epithelial cells: A mechanistic connection between the kidney defects in cpk mice and human ARPKD.
FASEB journal : official publication of the Federation of American Societies for Experimental BiologyRenal ciliopathies: promising drug targets and prospects for clinical trials.
Expert opinion on therapeutic targetsCystic Diseases of the Kidneys: From Bench to Bedside.
Indian journal of nephrologyShift from severe hypotension to salt-dependent hypertension in a child with autosomal recessive polycystic kidney disease after bilateral nephrectomies: a case report.
BMC nephrologyAutosomal dominant and autosomal recessive polycystic kidney disease: hypertension and secondary cardiovascular effect in children.
Frontiers in molecular biosciencesAN 84-YEAR-OLD PATIENT WITH CAROLI SYNDROME: WHAT IS THE PROGNOSIS OF THIS CONDITION?
European journal of case reports in internal medicineTRPV4 functional status in cystic cells regulates cystogenesis in autosomal recessive polycystic kidney disease during variations in dietary potassium.
Physiological reportsGenetic autopsy and genetic counseling for a case of fatal oligohydramnios due to de novo 17q12 deletion syndrome.
The journal of obstetrics and gynaecology researchAmeliorating liver disease in an autosomal recessive polycystic kidney disease mouse model.
American journal of physiology. Gastrointestinal and liver physiologyCompound heterozygosity of a de novo submicroscopic deletion and an inherited frameshift pathogenic variant in the PKHD1 gene in a fetus with bilaterally enlarged and echogenic kidneys, enlarged abdomen and oligohydramnios.
Clinical case reportsA Potential Therapy Using Antisense Oligonucleotides to Treat Autosomal Recessive Polycystic Kidney Disease.
Journal of clinical medicineAccuracy and processing time of kidney volume measurement methods in rodents polycystic kidney disease models: superiority of semiautomated kidney segmentation.
American journal of physiology. Renal physiologyDesign of two ongoing clinical trials of tolvaptan in the treatment of pediatric patients with autosomal recessive polycystic kidney disease.
BMC nephrologyPhosphomannomutase 2 (PMM2) variants leading to hyperinsulinism-polycystic kidney disease are associated with early-onset inflammatory bowel disease and gastric antral foveolar hyperplasia.
Human geneticsWhole-Exome Sequencing Revealed the Mutational Profiles of Primary Central Nervous System Lymphoma.
Clinical lymphoma, myeloma & leukemiaGlobal Transcriptomics of Congenital Hepatic Fibrosis in Autosomal Recessive Polycystic Kidney Disease using PCK rats.
bioRxiv : the preprint server for biologyTranscription factor Ap2b regulates the mouse autosomal recessive polycystic kidney disease genes, Pkhd1 and Cys1.
Frontiers in molecular biosciencesPolycystic Kidney Disease Drug Development: A Conference Report.
Kidney medicinePredominant Liver Cystic Disease in a New Heterozygotic PKHD1 Variant: A Case Report.
The American journal of case reportsReview of the Use of Animal Models of Human Polycystic Kidney Disease for the Evaluation of Experimental Therapeutic Modalities.
Journal of clinical medicineThe Clinical and Mutational Spectrum of 69 Turkish Children with Autosomal Recessive or Autosomal Dominant Polycystic Kidney Disease: A Multicenter Retrospective Cohort Study.
NephronSuspected Autosomal Recessive Polycystic Kidney Disease but Cerebellar Vermis Hypoplasia, Oligophrenia Ataxia, Coloboma, and Hepatic Fibrosis (COACH) Syndrome in Retrospect, A Delayed Diagnosis Aided by Genotyping and Reverse Phenotyping: A Case Report and A Review of the Literature.
NephronKidney concentrating capacity in children with autosomal recessive polycystic kidney disease is linked to glomerular filtration and hypertension.
Pediatric nephrology (Berlin, Germany)INDIAMAN-20 (INstant DIAgnosis of 20 Major ANomalies) protocol: application of IOTA diagnostic strategy to fetal anomalies.
Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and GynecologyIdentification and Characterization of Novel Mutations in Chronic Kidney Disease (CKD) and Autosomal Dominant Polycystic Kidney Disease (ADPKD) in Saudi Subjects by Whole-Exome Sequencing.
Medicina (Kaunas, Lithuania)Hyperinsulinemic Hypoglycemia Due to PMM2 Mutation in Two Siblings with Autosomal Recessive Polycystic Kidney Disease.
Pediatric reportsCystic Kidney Diseases That Require a Differential Diagnosis from Autosomal Dominant Polycystic Kidney Disease (ADPKD).
Journal of clinical medicineModulation of P2X4 receptor activity by ivermectin and 5-BDBD has no effect on the development of ARPKD in PCK rats.
Physiological reportsTwo cases of fetal hyperechogenic kidneys who had HNF1-β gene variation.
Clinical nephrologyTemporal Profile of Kynurenine Pathway Metabolites in a Rodent Model of Autosomal Recessive Polycystic Kidney Disease.
International journal of tryptophan research : IJTRPrenatal ultrasound in fetuses with polycystic kidney appearance - expanding the diagnostic algorithm.
Archives of gynecology and obstetrics[Clinical characteristics and genetic analysis of a child with autosomal recessive polycystic kidney disease].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsOrganoid-on-a-chip model of human ARPKD reveals mechanosensing pathomechanisms for drug discovery.
Science advancesPerspectives on Drug Development in Autosomal Recessive Polycystic Kidney Disease.
Clinical journal of the American Society of Nephrology : CJASNMolecular Diagnostics of Ciliopathies and Insights Into Novel Developments in Diagnosing Rare Diseases.
British journal of biomedical scienceEvaluation of galectin-3 and intestinal fatty acid binding protein as serum biomarkers in autosomal recessive polycystic kidney disease.
Journal of nephrologyCaroli's syndrome with autosomal recessive polycystic kidney disease on fetal MRI: A case report.
Congenital anomaliesThe Enigma of Clinical Heterogeneity Among Autosomal Recessive Polycystic Kidney Disease Siblings: PKHD1 Genotype Versus Other Genomic or Environmental Modifier.
Kidney international reportsPhenotypic Variability in Siblings With Autosomal Recessive Polycystic Kidney Disease.
Kidney international reportsPatient Selection for Renal Denervation in Hypertensive Patients: What Makes a Good Candidate?
Vascular health and risk managementGenetics, pathobiology and therapeutic opportunities of polycystic liver disease.
Nature reviews. Gastroenterology & hepatologyContributions of afferent and sympathetic renal nerves to cystogenesis and arterial pressure regulation in a preclinical model of autosomal recessive polycystic kidney disease.
American journal of physiology. Renal physiologyGeneration of induced pluripotent stem cells from peripheral blood mononuclear cells obtained from an adult with autosomal recessive polycystic kidney disease.
Stem cell researchPrimary URECs: a source to better understand the pathology of renal tubular epithelia in pediatric hereditary cystic kidney diseases.
Orphanet journal of rare diseasesDetection of DZIP1L mutations by whole-exome sequencing in consanguineous families with polycystic kidney disease.
Pediatric nephrology (Berlin, Germany)Ambulatory blood pressure and hypertension control in children with autosomal recessive polycystic kidney disease: clinical experience from two central European tertiary centres.
Journal of hypertensionThe genetics of Autosomal Recessive Polycystic Kidney Disease (ARPKD).
Biochimica et biophysica acta. Molecular basis of diseaseTargeted next-generation sequencing in a large series of fetuses with severe renal diseases.
Human mutationMosaic PKHD1 in Polycystic Kidneys Caused Aberrant Protein Expression in the Mitochondria and Lysosomes.
Frontiers in medicineEarly childhood height-adjusted total kidney volume as a risk marker of kidney survival in ARPKD.
Scientific reportsGeneration of an induced pluripotent stem cell line (DHMCi006-A) from a patient with autosomal recessive polycystic kidney disease (ARPKD) carrying a compound heterozygous missense mutation in the fibrocystin encoding PKHD1 gene.
Stem cell researchGeneration of an induced pluripotent stem cell line (DHMCi007-A) from a patient with autosomal recessive polycystic kidney disease (ARPKD) carrying a homozygous missense mutation in the fibrocystin-encoding PKHD1 gene.
Stem cell researchA human multi-lineage hepatic organoid model for liver fibrosis.
Nature communicationsClinical characteristics of Slovenian pediatric patients with autosomal recessive polycystic kidney disease.
Clinical nephrologyCan the Enhanced Liver Fibrosis Score Be Used to Diagnose Children With Liver Fibrosis?
Journal of pediatric gastroenterology and nutritionAutophagy-mediated reduction of miR-345 contributes to hepatic cystogenesis in polycystic liver disease.
JHEP reports : innovation in hepatologyClinical features of autosomal recessive polycystic kidney disease in the Japanese population and analysis of splicing in PKHD1 gene for determination of phenotypes.
Clinical and experimental nephrologyCystin genetic variants cause autosomal recessive polycystic kidney disease associated with altered Myc expression.
Scientific reportsAutosomal recessive polycystic kidney disease.
American journal of obstetrics and gynecologyDifferential Diagnosis and Prognosis of Fetuses with Bilateral Enlarged, Hyperechogenic Kidneys: Renal Volume and Amniotic Fluid Volume with Advancing Gestation.
Zeitschrift fur Geburtshilfe und Neonatologie[Clinical feature and genetic analysis of a fetus with autosomal recessive polycystic kidney disease].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsSystematic review on outcomes used in clinical research on autosomal recessive polycystic kidney disease-are patient-centered outcomes our blind spot?
Pediatric nephrology (Berlin, Germany)Therapeutic Potential for CFTR Correctors in Autosomal Recessive Polycystic Kidney Disease.
Cellular and molecular gastroenterology and hepatologyMolecular Pathophysiology of Autosomal Recessive Polycystic Kidney Disease.
International journal of molecular sciencesTwo-dimensional (2D) morphologic measurements can quantify the severity of liver disease in children with autosomal recessive polycystic kidney disease (ARPKD).
Abdominal radiology (New York)Rapid B1-Insensitive MR Fingerprinting for Quantitative Kidney Imaging.
Radiology[Diagnosis of a case of autosomal recessive polycystic kidney disease with combined prenatal imaging and genetic testing].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsInfant presenting with pyloric stenosis and autosomal recessive polycystic kidney disease at 36 weeks' postmenstrual age (PMA).
BMJ case reportsCongenital hepatic fibrosis: case report and review of literature.
The Pan African medical journalRisk factors for post-nephrectomy hypotension in pediatric patients.
Pediatric nephrology (Berlin, Germany)A case of 17q12 deletion syndrome that presented antenatally with markedly enlarged kidneys and clinically mimicked autosomal recessive polycystic kidney disease.
CEN case reportsRefining genotype-phenotype correlations in 304 patients with autosomal recessive polycystic kidney disease and PKHD1 gene variants.
Kidney internationalImaging manifestations of Caroli disease with autosomal recessive polycystic kidney disease: a case report and literature review.
BMC pregnancy and childbirthLoss of Cilia Does Not Slow Liver Disease Progression in Mouse Models of Autosomal Recessive Polycystic Kidney Disease.
Kidney360Fibrocystic liver disease: novel concepts and translational perspectives.
Translational gastroenterology and hepatologyIdentification of PKHD1 mutations in Brain, Breast and Rectal tumors by Next Generation DNA Sequencing.
The Gulf journal of oncologyEarly clinical management of autosomal recessive polycystic kidney disease.
Pediatric nephrology (Berlin, Germany)Polycystic liver disease genes: Practical considerations for genetic testing.
European journal of medical geneticsAdult Inactivation of the Recessive Polycystic Kidney Disease Gene Causes Polycystic Liver Disease.
Kidney360Autosomal Recessive Polycystic Kidney Disease-The Clinical Aspects and Diagnostic Challenges.
Journal of pediatric geneticsPredictors of progression in autosomal dominant and autosomal recessive polycystic kidney disease.
Pediatric nephrology (Berlin, Germany)Multiple Cerebral Aneurysms in an Adult With Autosomal Recessive Polycystic Kidney Disease.
Kidney international reportsDiagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021.
American journal of kidney diseases : the official journal of the National Kidney FoundationReverse Phenotyping Maternal Cystic Kidney Disease by Diagnosis in a Newborn: Case Report and Literature Review on Neonatal Cystic Kidney Diseases.
Acta medica LituanicaPlasticity of distal nephron epithelia from human kidney organoids enables the induction of ureteric tip and stalk.
Cell stem cell[Infantile arterial hypertension: A diagnostic challenge in paediatrics].
Anales de pediatriaExploring the Spectrum of Kidney Ciliopathies.
Diagnostics (Basel, Switzerland)Etiologies and outcomes of prenatally diagnosed hyperechogenic kidneys.
Prenatal diagnosisOccurrence of Portal Hypertension and Its Clinical Course in Patients With Molecularly Confirmed Autosomal Recessive Polycystic Kidney Disease (ARPKD).
Frontiers in pediatricsCaroli's Syndrome: An Early Presentation.
CureusIntrahepatic bile ductal ectasia in autosomal recessive polycystic kidney disease evaluated by fetal magnetic resonance imaging: a more frequent complication.
The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal ObstetriciansLong-term kidney and liver outcome in 50 children with autosomal recessive polycystic kidney disease.
Pediatric nephrology (Berlin, Germany)Combined liver and kidney transplantation in children and long-term outcome.
World journal of transplantationPrevalence, risk factors and disease knowledge of polycystic kidney disease in Pakistan.
International journal of immunopathology and pharmacologyPossible PKHD1 Hot-spot Mutations Related to Early Kidney Function Failure or Hepatofibrosis in Chinese Children with ARPKD: A Retrospective Single Center Cohort Study and Literature Review.
Current medical scienceThe carboxy-terminus of the human ARPKD protein fibrocystin can control STAT3 signalling by regulating SRC-activation.
Journal of cellular and molecular medicineAutosomal recessive polycystic kidney disease: case report of a newborn with rare PKHD1 mutation, rapid renal enlargement and early fatal outcome.
Italian journal of pediatricsUse of patient derived urine renal epithelial cells to confirm pathogenicity of PKHD1 alleles.
BMC nephrologyCollecting duct cells show differential retinoic acid responses to acute versus chronic kidney injury stimuli.
Scientific reportsSevere neurological outcomes after very early bilateral nephrectomies in patients with autosomal recessive polycystic kidney disease (ARPKD).
Scientific reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença renal policística autossômica recessiva.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença renal policística autossômica recessiva
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Deciphering the Impact of RAC1-SPTAN1 in ARPKD Cystogenesis Using Multifaceted Models.
- Congenital Hepatic Fibrosis and/or Autosomal Recessive Polycystic Kidney Disease: A Single-center Experience.
- Identification of PDIA6 Mutation in a Case of Autosomal Recessive Polycystic Kidney Disease: A Case Report and Review of Literature.
- Advances in pediatric kidney diffusion tensor imaging: diagnostic and functional applications.
- Caroli disease associated with autosomal recessive polycystic kidney disease: CT imaging features of a case report.
- [Research advances in the diagnosis and treatment of Polycystic kidney disease].
- Pegcetacoplan-induced remission in pediatric immune-complex membranoproliferative glomerulonephritis with comorbid autosomal recessive polycystic kidney disease: a case report.
- Monogenic Etiologies of Kidney Cysts in the Pediatric Population: An Observational Cohort Study.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:731(Orphanet)
- MONDO:0009889(MONDO)
- GARD:8378(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3395618(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
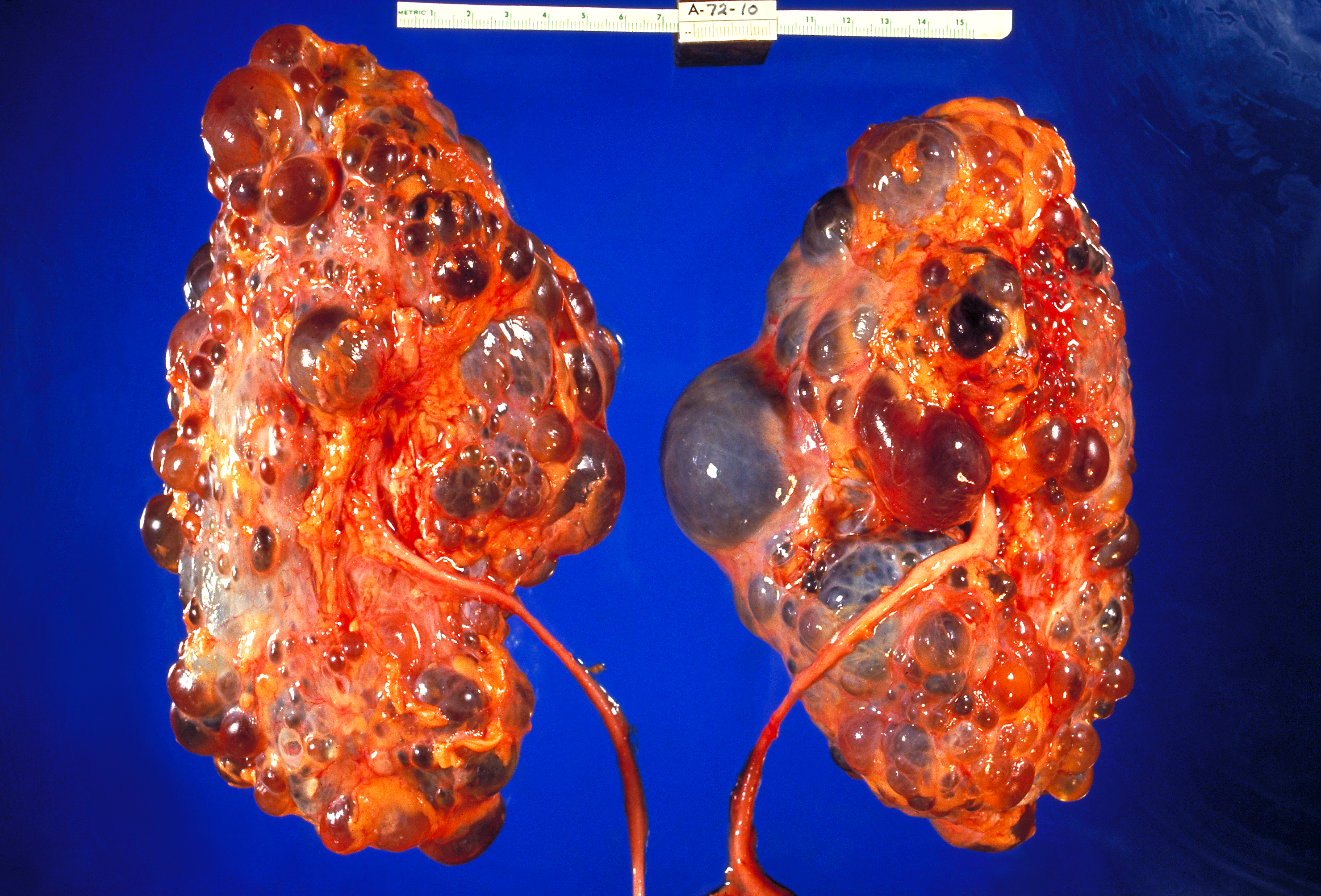
Doença renal policística autossômica recessiva
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos (literatura)
- fonte: Orphanet
- Ensaios clínicos
- fonte: ClinicalTrials.gov