Uma doença genética rara e fatal que provoca múltiplas anomalias (problemas de nascença). É caracterizada por uma combinação de três problemas principais: uma má-formação cerebral, geralmente uma protuberância na parte de trás da cabeça (encefalocele occipital); rins grandes e com vários cistos (bolsinhas de líquido); e dedos a mais nas mãos ou nos pés (polidactilia). Anormalidades associadas também podem incluir lábio leporino ou fenda palatina (no céu da boca), problemas no coração e nos órgãos genitais, outras má-formações no sistema nervoso central (cérebro e medula espinhal), fibrose (cicatrização) no fígado e alterações no desenvolvimento dos ossos.
Introdução
O que você precisa saber de cara
Uma doença genética rara e fatal que provoca múltiplas anomalias (problemas de nascença). É caracterizada por uma combinação de três problemas principais: uma má-formação cerebral, geralmente uma protuberância na parte de trás da cabeça (encefalocele occipital); rins grandes e com vários cistos (bolsinhas de líquido); e dedos a mais nas mãos ou nos pés (polidactilia). Anormalidades associadas também podem incluir lábio leporino ou fenda palatina (no céu da boca), problemas no coração e nos órgãos genitais, outras má-formações no sistema nervoso central (cérebro e medula espinhal), fibrose (cicatrização) no fígado e alterações no desenvolvimento dos ossos.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 81 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 194 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
19 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Component of the tectonic-like complex, a complex localized at the transition zone of primary cilia and acting as a barrier that prevents diffusion of transmembrane proteins between the cilia and plasma membranes. Required for ciliogenesis and sonic hedgehog/SHH signaling (By similarity)
Cytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, cilium axoneme
Meckel syndrome 9
A disorder characterized by a combination of renal cysts and variably associated features including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia and cysts, and polydactyly.
Microtubule motor protein that binds to microtubules with high affinity through each tubulin heterodimer and has an ATPase activity (By similarity). Plays a role in many processes like cell division, cytokinesis and also in cell proliferation and apoptosis (PubMed:16648480, PubMed:24784001). During cytokinesis, targets to central spindle and midbody through its interaction with PRC1 and CIT respectively (PubMed:16431929). Regulates cell growth through regulation of cell cycle progression and cyt
NucleusCytoplasmCytoplasm, cytoskeleton, spindleMidbody
Meckel syndrome 12
A form of Meckel syndrome, a disorder characterized by a combination of renal cysts and variably associated features including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia and cysts, and polydactyly.
Required for normal ciliary development and function. Inhibits disheveled-1-induced canonical Wnt-signaling activity and may also play a role in the control of non-canonical Wnt signaling which regulates planar cell polarity. Probably acts as a molecular switch between different Wnt signaling pathways. Required for proper convergent extension cell movements
Cell projection, cilium
Nephronophthisis 3
An autosomal recessive disorder resulting in end-stage renal disease. It is characterized by polyuria, polydipsia, anemia. Onset of terminal renal failure occurr significantly later (median age, 19 years) than in juvenile nephronophthisis. Renal pathology is characterized by alterations of tubular basement membranes, tubular atrophy and dilation, sclerosing tubulointerstitial nephropathy, and renal cyst development predominantly at the corticomedullary junction.
Essential for primary ciliogenesis and embryonic development, facilitating the activation of Hedgehog (Hh) signaling pathway. Disrupts the interaction of GLI2 and GLI3 with the negative regulator SUFU. Inhibiting SUFU's interaction with GLI2 promotes the entry of GLI2 into the nucleus, allowing it to activate Hh target gene expression. Disrupting SUFU's interaction with GLI3 prevents its conversion into the repressor form, leading to increased nuclear GLI3 and enhanced Hh signaling. Required for
MembraneCytoplasm, cytoskeleton, cilium basal body
Joubert syndrome 2
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease.
Required for ciliary structure and function. Part of the tectonic-like complex which is required for tissue-specific ciliogenesis and may regulate ciliary membrane composition (By similarity). Involved in centrosome migration to the apical cell surface during early ciliogenesis. Involved in the regulation of cilia length and appropriate number through the control of centrosome duplication. Is a key regulator of stereociliary bundle orientation (By similarity). Required for epithelial cell branch
Cell membraneEndoplasmic reticulum membraneCell projection, ciliumCytoplasm, cytoskeleton, cilium basal body
Acts as a positive regulator of ciliary hedgehog signaling (By similarity). Involved in ciliogenesis (PubMed:27894351)
Cell projection, cilium membrane
Meckel syndrome 14
A form of Meckel syndrome, an autosomal recessive disorder characterized by a combination of renal cysts and variably associated features including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia and cysts, and polydactyly. Death occurs in the prenatal or perinatal period.
May play a role in cell-cycle-dependent microtubule organization
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, spindleCytoplasm, cytoskeleton, spindle poleCell projection, cilium
Joubert syndrome 21
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy, renal disease, liver fibrosis, and polydactyly.
Component of the tectonic-like complex, a complex localized at the transition zone of primary cilia and acting as a barrier that prevents diffusion of transmembrane proteins between the cilia and plasma membranes. Regulator of Hedgehog (Hh), required for both activation and inhibition of the Hh pathway in the patterning of the neural tube. During neural tube development, it is required for formation of the most ventral cell types and for full Hh pathway activation. Functions in Hh signal transdu
Cytoplasm, cytoskeleton, cilium basal bodySecreted
Joubert syndrome 13
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease.
Involved in early and late steps in cilia formation. Its association with CCP110 is required for inhibition of primary cilia formation by CCP110 (PubMed:18694559). May play a role in early ciliogenesis in the disappearance of centriolar satellites and in the transition of primary ciliar vesicles (PCVs) to capped ciliary vesicles (CCVs). Required for the centrosomal recruitment of RAB8A and for the targeting of centriole satellite proteins to centrosomes such as of PCM1 (PubMed:24421332). Require
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriolar satelliteNucleusCell projection, ciliumCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centrioleCytoplasmic vesicle
Joubert syndrome 5
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease. Joubert syndrome type 5 shares the neurologic and neuroradiologic features of Joubert syndrome together with severe retinal dystrophy and/or progressive renal failure characterized by nephronophthisis.
Part of the tectonic-like complex which is required for tissue-specific ciliogenesis and may regulate ciliary membrane composition (By similarity). May be involved in apoptosis regulation. Necessary for signal transduction through the sonic hedgehog (Shh) signaling pathway
Membrane
Orofaciodigital syndrome 4
A form of orofaciodigital syndrome, a group of heterogeneous disorders characterized by malformations of the oral cavity, face and digits, and associated phenotypic abnormalities that lead to the delineation of various subtypes. OFD4 patients have tongue nodules, multiple frenulae, broad flat nose, hypertelorism, and short rib polydactyly with tibial dysplasia (Majewski syndrome). The presence of severe tibial aplasia differentiates OFD4 from OFD1. Additional features of cystic dysplastic kidneys and brain malformation, including occipital encephalocele, are observed in severely affected patients.
Negatively regulates signaling through the G-protein coupled thromboxane A2 receptor (TBXA2R) (PubMed:19464661). May be involved in mechanisms like programmed cell death, craniofacial development, patterning of the limbs, and formation of the left-right axis (By similarity). Involved in the organization of apical junctions; the function is proposed to implicate a NPHP1-4-8 module. Does not seem to be strictly required for ciliogenesis (PubMed:19464661). Involved in establishment of planar cell p
CytoplasmCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, cilium axonemeCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCell junction, tight junction
Transmembrane component of the tectonic-like complex, a complex localized at the transition zone of primary cilia and acting as a barrier that prevents diffusion of transmembrane proteins between the cilia and plasma membranes. Required for ciliogenesis and sonic hedgehog/SHH signaling (By similarity)
Cell projection, cilium membrane
Joubert syndrome 20
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease.
Component of the transition zone in primary cilia. Required for ciliogenesis
MembraneCell projection, cilium
Joubert syndrome 14
An autosomal recessive disorder characterized by severe intellectual disability, hypotonia, breathing abnormalities in infancy, and dysmorphic facial features. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include renal disease, abnormal eye movements, and postaxial polydactyly.
Plays a role in cilia formation and embryonic patterning. Requires for normal Sonic hedgehog (Shh) signaling in the neural tube and acts in combination with GLI2 and GLI3 to pattern ventral and intermediate neuronal cell types (By similarity). During ciliogenesis regulates the ciliary transition zone localization of some MKS complex proteins (PubMed:26518474)
MembraneCell projection, cilium
Meckel syndrome 13
A form of Meckel syndrome, a disorder characterized by a combination of renal cysts and variably associated features including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia and cysts, and polydactyly.
Component of the tectonic-like complex, a complex localized at the transition zone of primary cilia and acting as a barrier that prevents diffusion of transmembrane proteins between the cilia and plasma membranes. Involved in centrosome migration to the apical cell surface during early ciliogenesis. Required for ciliary structure and function, including a role in regulating length and appropriate number through modulating centrosome duplication. Required for cell branching morphology
Cytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Meckel syndrome 1
A disorder characterized by a combination of renal cysts and variably associated features including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia and cysts, and polydactyly.
Component of the tectonic-like complex, a complex localized at the transition zone of primary cilia and acting as a barrier that prevents diffusion of transmembrane proteins between the cilia and plasma membranes. Required for ciliogenesis and sonic hedgehog/SHH signaling (By similarity)
CytoplasmCytoplasm, cytoskeleton, cilium basal body
Meckel syndrome 6
A disorder characterized by a combination of renal cysts and variably associated features including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia and cysts, and polydactyly.
May function as scaffolding protein. Required for normal location of RPGR at the connecting cilium of photoreceptor cells. Required for normal disk morphogenesis and disk organization in the outer segment of photoreceptor cells and for survival of photoreceptor cells
Cell projection, cilium
Leber congenital amaurosis 6
A severe dystrophy of the retina, typically becoming evident in the first years of life. Visual function is usually poor and often accompanied by nystagmus, sluggish or near-absent pupillary responses, photophobia, high hyperopia and keratoconus.
Component of the tectonic-like complex, a complex localized at the transition zone of primary cilia and acting as a barrier that prevents diffusion of transmembrane proteins between the cilia and plasma membranes. Required for hedgehog signaling transduction (By similarity)
MembraneCytoplasm, cytoskeleton, cilium basal body
Meckel syndrome 8
A disorder characterized by a combination of renal cysts and variably associated features including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia and cysts, and polydactyly.
Component of the tectonic-like complex, a complex localized at the transition zone of primary cilia and acting as a barrier that prevents diffusion of transmembrane proteins between the cilia and plasma membranes
Cytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, cilium axonemeNucleus
Meckel syndrome 10
A disorder characterized by a combination of renal cysts and variably associated features including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia and cysts, and polydactyly.
Variantes genéticas (ClinVar)
483 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 3.167 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
20 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Meckel
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Missense Variants in the Second Transmembrane Domain of TMEM17 Disrupt Its Stability and Function and Lead to a Wide Phenotypic Spectrum of Ciliopathies.
Ciliopathies are rare genetic disorders characterized by significant genetic and phenotypic variability. Over 140 proteins localized to primary cilia, which are sensory organelles essential for vertebrate development, are implicated. TMEM17 encodes a transmembrane protein at the ciliary transition zone and was previously proposed as a potential ciliopathy gene, based on reports of individuals from two families with orofaciodigital syndrome type 6 (OFD6) and Joubert syndrome (JS). Here, we report two unrelated fetuses with occipital encephalocele, polydactyly, and kidney cysts, in whom exome sequencing identified a founder homozygous missense variant (Arg94Trp) in TMEM17, affecting a highly conserved residue. This expands the TMEM17-associated phenotypic spectrum to include Meckel syndrome (MKS). Comprehensive functional analyses of all known TMEM17 variants, using patient tissues/cells and a C. elegans model system, demonstrate a loss-of-function mechanism. Our study reveals severe functional consequences, including TMEM17 destabilization and mislocalization, anomalies in cilium composition and function, and abrogation of Sonic Hedgehog signaling. These experiments confirm the pathogenicity of all TMEM17 variants and underscore its essential role at the ciliary transition zone. Collectively, our findings establish TMEM17 as a bona fide ciliopathy gene, associated with a wide phenotypic spectrum ranging from viable syndromes (OFD6 and JS) to a fetal-lethal condition (MKS).
CRISPR-Cas9-Generated TXNDC15 c.560delA Homozygous Mouse Model Exhibits Meckel-Gruber Syndrome Phenotype.
To determine whether TXNDC15 variation causes Meckel-Gruber syndrome (MKS), we assessed the pathogenicity of the frameshift variant c.560delA. A CRISPR-Cas9 generated mouse model carrying the equivalent Txndc15 c.512delA mutation was analyzed at embryonic day 15.5. Homozygous Txndc15mt/mt embryos displayed the complete MKS phenotype-fetal lethality, exencephaly, omphalocele, post-axial polydactyly, and polycystic kidneys-together with markedly reduced TXNDC15 protein in brain, liver, and kidney. These findings confirm TXNDC15 as a bona fide MKS disease gene.
Ciliopathy-related B9 protein complex regulates ciliary axonemal microtubule posttranslational modifications and initiation of ciliogenesis.
Ciliary dysfunction results in multiorgan developmental diseases, collectively known as ciliopathies. The B9D1-B9D2-MKS1protein complex maintains the gatekeeper function at the ciliary transition zone (TZ). However, the function of B9 proteins and the mechanisms underlying why different variants in the same B9 gene cause different ciliopathies are not fully understood. Here, we investigated the function of B9 proteins and revealed 2 critical functions. First, the B9 complex interacted with and anchored TMEM67 to the TZ membrane. Disruption of the B9-TMEM67 complex reduced posttranslational modifications of axonemal microtubules due to deregulation of tubulin-modifying enzymes within cilia. Second, B9 proteins localized to centrioles prior to ciliogenesis, where they facilitated the initiation of ciliogenesis. In addition, we identified B9D2 variants in a cohort of patients with Joubert syndrome. We found that Joubert syndrome-associated B9D2 variants primarily affected axonemal microtubule modifications without disrupting ciliogenesis, whereas the Meckel syndrome-associated B9D2 variant disrupted both ciliogenesis and axonemal microtubule modifications. Thus, besides its role as a gatekeeper for ciliary membrane proteins, the B9 complex also controls axonemal microtubule posttranslational modifications and early stages of ciliogenesis, providing insights into the distinct pathologies arising from different variants of the same gene.
Biallelic Variants in TMEM17 Cause Meckel-Gruber Syndrome Within the Ciliopathy Spectrum.
TMEM17 encodes a transition zone protein essential for ciliary function. Three cases with homozygous variants in TMEM17 in primary ciliopathies (Joubert and Oral-Facial-Digital syndrome) have been reported. We investigated whether biallelic TMEM17 variants contribute to primary ciliopathies. We queried our Biodatabank and evaluated the gene-disease relationship (GDR) according to the ClinGen recommendations. Four unrelated patients (four families) were identified with a clinical diagnosis of Meckel-Gruber syndrome (MGS) and novel homozygous variants: NM_198276.3:c.4del p.(Glu2Serfs*58); NM_198276.3:c.366dup p.(Pro123Thrfs*9); and NM_198276.3:c.368C>G p.(Pro123Arg). A fifth family lost three foetuses with MGS phenotype, both parents are heterozygote carriers (NM_198276.3:c.4del p.(Glu2Serfs*58)) but biological material from the foetuses was not available. The cases in this study had a severe prenatal phenotype, including encephalocele, polycystic kidney dysplasia, and polydactyly, leading to early lethality. This study strengthens the gene-disease association of TMEM17, upgrading it from "limited" to "moderate." We expand the phenotypic spectrum, ranging from MGS-with prenatal onset and early lethality-to Oral-Facial-Digital and Joubert syndromes. Our findings indicate that loss-of-function variants may underlie the most severe TMEM17 ciliopathy manifestations, suggesting a potential genotype-phenotype correlation.
Cleavage of the Meckel-Gruber syndrome protein TMEM67 by ADAMTS9 uncouples Wnt signaling and ciliogenesis.
TMEM67 mutations cause Meckel-Gruber syndrome and other related ciliopathies. TMEM67 is involved in both ciliary transition zone assembly, and non-canonical Wnt signaling mediated by its extracellular domain. How TMEM67 performs these two separate functions is not known. We identify a cleavage motif in the extracellular domain of TMEM67 cleaved by the extracellular matrix metalloproteinase ADAMTS9. This cleavage regulates the abundance of two functional forms: a C-terminal portion which localizes to the ciliary transition zone regulating ciliogenesis, and a non-cleaved form which regulates Wnt signaling. By characterizing three TMEM67 ciliopathy patient variants within the cleavage motif utilizing mammalian cell culture and C. elegans, we show the cleavage motif is essential for cilia structure and function, highlighting its clinical significance. We generated a non-cleavable TMEM67 mouse model which develop severe ciliopathies phenocopying Tmem67-/- mice, but in contrast, transduces normal Wnt signaling, substantiating the existence of two functional forms of TMEM67.
Publicações recentes
Novel Compound Heterozygous Variants in the TCTN2 Gene Causing Meckel-Gruber Syndrome 8 in a Non-Consanguineous Chinese Family.
Ciliopathy-related B9 protein complex regulates ciliary axonemal microtubule posttranslational modifications and initiation of ciliogenesis.
Compound heterozygous missense and intronic variants in B9D1 contribute to a recurrent Meckel syndrome pedigree.
Missense Variants in the Second Transmembrane Domain of TMEM17 Disrupt Its Stability and Function and Lead to a Wide Phenotypic Spectrum of Ciliopathies.
First-trimester diagnosis of Meckel syndrome by ultrasonography with suspected mutation of CC2D2A: a case description.
📚 EuropePMC133 artigos no totalmostrando 128
Meckel-Gruber syndrome: a rare and fatal congenital disorder (case report).
The Pan African medical journalCRISPR-Cas9-Generated TXNDC15 c.560delA Homozygous Mouse Model Exhibits Meckel-Gruber Syndrome Phenotype.
Genesis (New York, N.Y. : 2000)Novel Compound Heterozygous Variants in the TCTN2 Gene Causing Meckel-Gruber Syndrome 8 in a Non-Consanguineous Chinese Family.
Molecular genetics & genomic medicineCiliopathy-related B9 protein complex regulates ciliary axonemal microtubule posttranslational modifications and initiation of ciliogenesis.
The Journal of clinical investigationBiallelic Variants in TMEM17 Cause Meckel-Gruber Syndrome Within the Ciliopathy Spectrum.
Clinical geneticsCompound heterozygous missense and intronic variants in B9D1 contribute to a recurrent Meckel syndrome pedigree.
Frontiers in geneticsMissense Variants in the Second Transmembrane Domain of TMEM17 Disrupt Its Stability and Function and Lead to a Wide Phenotypic Spectrum of Ciliopathies.
Clinical geneticsInvestigating the Role of B9D1 in Meckel-Gruber Syndrome: A Case Report and Comprehensive Literature Review.
GenesCleavage of the Meckel-Gruber syndrome protein TMEM67 by ADAMTS9 uncouples Wnt signaling and ciliogenesis.
Nature communicationsFirst-trimester diagnosis of Meckel syndrome by ultrasonography with suspected mutation of CC2D2A: a case description.
Quantitative imaging in medicine and surgerySevere Joubert syndrome in family with homozygous POC1B p.Arg106Pro variant is due to a co-inherited deep-intronic mutation in the neighboring CEP290 gene.
HGG advancesApproaches to Evaluate Whole Exome Sequencing Data That Incorporate Genetic Intolerance Scores for Congenital Anomalies, Including Intronic Regions Adjacent to Exons.
Molecular genetics & genomic medicineRegulation of INPP5E in Ciliogenesis, Development, and Disease.
International journal of biological sciencesA Missense Variant in KIF14 Results in Two Gene Isoforms by Affecting Normal Gene Splicing.
Prenatal diagnosisVariant Spectrum of Renal Ciliopathies in Turkish Cohort and Genotype-Phenotype Association Specifically in Autosomal Dominant Polycystic Kidney Disease.
Clinical geneticsTXNDC15, an ER-localized thioredoxin-like transmembrane protein, contributes to ciliary transition zone integrity.
Journal of cell scienceNew functions of B9D2 in tight junctions and epithelial polarity.
Scientific reportsA Rare Case of Meckel-Gruber Syndrome with Congenital Intestinal Atresia and Abdominal Pseudocyst Clinic.
Fetal and pediatric pathologyBiallelic TXNDC15 variants associated with Joubert syndrome-related molar tooth sign and forebrain malformation.
Journal of human geneticsExpanding the phenotypic spectrum of CC2D2A-related ciliopathies: a rare homozygous nonsense variant in a patient with suspected nephronophthisis.
European journal of human genetics : EJHGTwo novel TMEM67 variations in a Chinese family with recurrent pregnancy loss: a case report.
BMC medical genomicsDefects in diffusion barrier function of ciliary transition zone caused by ciliopathy variations of TMEM218.
Human molecular geneticsNeurosurgical intervention for the Meckel-Gruber Syndrome: A systematic review.
Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery[Genetic analysis of a fetus with Meckel syndrome due to variants of TMEM67 gene].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsJoubert syndrome causing mutation in C2 domain of CC2D2A affects structural integrity of cilia and cellular signaling molecules.
Experimental brain researchNovel homozygous mutations in TXNDC15 causing Meckel syndrome.
Molecular genetics & genomic medicineFirst preimplantation genetic testing case of Meckel syndrome with a novel homozygous TXNDC15 variant in a non-consanguineous Chinese family.
Molecular genetics & genomic medicineCEP55-associated lethal fetal syndrome: a case report of a Chinese family.
Frontiers in geneticsIdentification of novel TMEM231 gene splice variants and pathological findings in a fetus with Meckel Syndrome.
Frontiers in genetics[Analysis of a Chinese pedigree affected with Meckel syndrome due to variants of TMEM67 gene].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsMeckel-Gruber syndrome together with Dandy-Walker malformation: an atypical case report of a 2nd recurrence in a consanguine marriage.
Child's nervous system : ChNS : official journal of the International Society for Pediatric NeurosurgeryIdentification of pathogenic deep intronic variant and exonic LINE-1 insertion in a patient with Meckel syndrome.
Annals of human geneticsJoubert syndrome: Molecular basis and treatment.
Journal of mother and childVariable phenotypes and penetrance between and within different zebrafish ciliary transition zone mutants.
Disease models & mechanismsPrenatal ultrasound in fetuses with polycystic kidney appearance - expanding the diagnostic algorithm.
Archives of gynecology and obstetricsThe Joubert-Meckel-Nephronophthisis Spectrum of Ciliopathies.
Annual review of genomics and human geneticsCase Report: Preimplantation Genetic Testing for Meckel Syndrome Induced by Novel Compound Heterozygous Mutations of MKS1.
Frontiers in geneticsEvaluation of novel compound variants of CEP290 in prenatally suspected case of Meckel syndrome through whole exome sequencing.
Molecular genetics & genomic medicineIdentification of Pathogenic Variants in RPGRIP1L with Meckel Syndrome and Preimplantation Genetic Testing in a Chinese Family.
Reproductive sciences (Thousand Oaks, Calif.)The ciliary transition zone protein TMEM218 synergistically interacts with the NPHP module and its reduced dosage leads to a wide range of syndromic ciliopathies.
Human molecular geneticsA SOX3 duplication and lumbosacral spina bifida in three generations.
American journal of medical genetics. Part AThree Novel Variants of CEP290 and CC2D2DA and a Link Between ZNF77 and SHH Signaling Pathway Are Found in Two Meckel-Gruber Syndrome Fetuses.
Reproductive sciences (Thousand Oaks, Calif.)[Genetic testing and prenatal diagnosis for a Chinese pedigree affected with Meckel-Gruber syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsMeckel Gruber and Joubert Syndrome Diagnosed Prenatally: Allelism between the Two Ciliopathies, Complexities of Mutation Types and Digenic Inheritance.
Fetal and pediatric pathologyPrenatal Exome Sequencing in Recurrent Fetal Structural Anomalies: Systematic Review and Meta-Analysis.
Journal of clinical medicineFetal ciliopathies: a retrospective observational single-center study.
Archives of gynecology and obstetricsHK1 haemolytic anaemia in association with a neurological phenotype and co-existing CEP290 Meckel-Gruber in a Romani family.
Clinical geneticsPrenatal Versus Postnatal Diagnosis of Meckel-Gruber and Joubert Syndrome in Patients with TMEM67 Mutations.
GenesMeckel-Gruber Syndrome: Clinical and Molecular Genetic Profiles in Two Fetuses and Review of the Current Literature.
Genetic testing and molecular biomarkersTMEM218 dysfunction causes ciliopathies, including Joubert and Meckel syndromes.
HGG advancesA founder mutation in TCTN2 causes Meckel-Gruber syndrome type 8 among Jews of Ethiopian and Yemenite origin.
American journal of medical genetics. Part Amtor Haploinsufficiency Ameliorates Renal Cysts and Cilia Abnormality in Adult Zebrafish tmem67 Mutants.
Journal of the American Society of Nephrology : JASNUpdate of genetic variants in CEP120 and CC2D2A-With an emphasis on genotype-phenotype correlations, tissue specific transcripts and exploring mutation specific exon skipping therapies.
Molecular genetics & genomic medicineCEP55 promotes cilia disassembly through stabilizing Aurora A kinase.
The Journal of cell biologyCilia, ciliopathies and hedgehog-related forebrain developmental disorders.
Neurobiology of diseaseInterpreting the pathogenicity of Joubert syndrome missense variants in Caenorhabditis elegans.
Disease models & mechanismsDandy-Walker Malformation.
American journal of obstetrics and gynecologyCiliopathies and the Kidney: A Review.
American journal of kidney diseases : the official journal of the National Kidney FoundationFormation of the B9-domain protein complex MKS1-B9D2-B9D1 is essential as a diffusion barrier for ciliary membrane proteins.
Molecular biology of the cellTMEM216 Deletion Causes Mislocalization of Cone Opsin and Rhodopsin and Photoreceptor Degeneration in Zebrafish.
Investigative ophthalmology & visual scienceTwo novel TCTN2 mutations cause Meckel-Gruber syndrome.
Journal of human geneticsTwo novel B9D1 variants causing Joubert syndrome: Utility of mRNA and splicing studies.
European journal of medical geneticsDNA Variant in the RPGRIP1L Gene Influences Alternative Splicing.
Molecular neuropsychiatryRenal-Hepatic-Pancreatic Dysplasia: An Ultra-Rare Ciliopathy with a Novel NPHP3 Genotype.
Journal of pediatric geneticsPrenatal diagnosis and clinical significance of cephalocele-A single institution experience and literature review.
Prenatal diagnosisWhole exome sequencing identified a homozygous novel variant in CEP290 gene causes Meckel syndrome.
Journal of cellular and molecular medicineCongenital talipes equinovarus (clubfoot).
American journal of obstetrics and gynecologyThe molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity.
Translational science of rare diseasesLong-read nanopore sequencing resolves a TMEM231 gene conversion event causing Meckel-Gruber syndrome.
Human mutationMeckel syndrome: Clinical and mutation profile in six fetuses.
Clinical geneticsMeckel's diverticulum and the eponymous legend.
The journal of trauma and acute care surgeryMeckel-Gruber Syndrome: A Case Who Lived for 5 Months.
Pediatric neurosurgeryCo-Occurrence of Leber Congenital Amaurosis and Meckel Syndrome Type 1 in a Fetus: Is There a Lesson to Be Learned?
Molecular syndromologyA prenatally diagnosed case of Meckel-Gruber syndrome with novel compound heterozygous pathogenic variants in the TXNDC15 gene.
Molecular genetics & genomic medicineHydrocephalus in a rat model of Meckel Gruber syndrome with a TMEM67 mutation.
Scientific reportsMks6 mutations reveal tissue- and cell type-specific roles for the cilia transition zone.
FASEB journal : official publication of the Federation of American Societies for Experimental BiologySuper-Resolution Imaging Reveals TCTN2 Depletion-Induced IFT88 Lumen Leakage and Ciliary Weakening.
Biophysical journalBasal exon skipping and nonsense-associated altered splicing allows bypassing complete CEP290 loss-of-function in individuals with unusually mild retinal disease.
Human molecular geneticsNew Insights into Cystic Kidney Diseases.
Contributions to nephrologyHistory and highlights of the teratological collection in the Museum Anatomicum of Leiden University, The Netherlands.
American journal of medical genetics. Part AA rare case of Meckel-Gruber syndrome.
Romanian journal of morphology and embryology = Revue roumaine de morphologie et embryologieSuper-resolution microscopy reveals that disruption of ciliary transition-zone architecture causes Joubert syndrome.
Nature cell biologyThe First Reported Case of Meckel-Gruber Syndrome Associated With Abnormal Karyotype Mosaic Trisomy 17.
Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology SocietyEXPANDED PHENOTYPE OF TMEM67 GENE MUTATION (CASE REPORT).
Georgian medical newsIsolated congenital hepatic fibrosis associated with TMEM67 mutations: report of a new genotype-phenotype relationship.
Clinical case reportsExpanding the allelic disorders linked to TCTN1 to include Varadi syndrome (Orofaciodigital syndrome type VI).
American journal of medical genetics. Part AUniparental disomy as an unexpected cause of Meckel-Gruber syndrome: report of a case.
Pediatric nephrology (Berlin, Germany)Meckel-Gruber syndrome: ultrasonographic and fetal autopsy correlation.
Journal of ultrasoundAn ovine hepatorenal fibrocystic model of a Meckel-like syndrome associated with dysmorphic primary cilia and TMEM67 mutations.
Scientific reportsDiagnostic use of computational retrotransposon detection: Successful definition of pathogenetic mechanism in a ciliopathy phenotype.
American journal of medical genetics. Part AA nonsense mutation in CEP55 defines a new locus for a Meckel-like syndrome, an autosomal recessive lethal fetal ciliopathy.
Clinical geneticsThe Meckel syndrome- associated protein MKS1 functionally interacts with components of the BBSome and IFT complexes to mediate ciliary trafficking and hedgehog signaling.
PloS oneFifteen years of research on oral-facial-digital syndromes: from 1 to 16 causal genes.
Journal of medical geneticsPrenatal ultrasound, genotype, and outcome in a large cohort of prenatally affected patients with autosomal-recessive polycystic kidney disease and other hereditary cystic kidney diseases.
Archives of gynecology and obstetrics[Meckel Gruber syndrome: about a rare case].
The Pan African medical journalCharacterizing the morbid genome of ciliopathies.
Genome biologyMotile and non-motile cilia in human pathology: from function to phenotypes.
The Journal of pathologyCilium transition zone proteome reveals compartmentalization and differential dynamics of ciliopathy complexes.
Proceedings of the National Academy of Sciences of the United States of AmericaTMEM231 Gene Conversion Associated with Joubert and Meckel-Gruber Syndromes in the Same Family.
Human mutationAnnals of morphology fields and prepatterns. Editorial Festschrift for John C. Carey, MD, MPH.
American journal of medical genetics. Part A[Prenatal diagnosis of Meckel-Gruber syndrome. Case report and literature review].
Ginecologia y obstetricia de MexicoMutations in CEP120 cause Joubert syndrome as well as complex ciliopathy phenotypes.
Journal of medical geneticsUpdate on oral-facial-digital syndromes (OFDS).
CiliaPrimary cilia maintain corneal epithelial homeostasis by regulation of the Notch signaling pathway.
Development (Cambridge, England)Clinical utility gene card for: Meckel syndrome - update 2016.
European journal of human genetics : EJHGMKS5 and CEP290 Dependent Assembly Pathway of the Ciliary Transition Zone.
PLoS biologyAn unusual case of Meckel-Gruber syndrome (MKS) associated with visceroatrial heterotaxy and facial anomalies.
Journal of obstetrics and gynaecology : the journal of the Institute of Obstetrics and GynaecologyMechanism of pancreatic and liver malformations in human fetuses with short-rib polydactyly syndrome.
Birth defects research. Part A, Clinical and molecular teratologyA Screen for Modifiers of Cilia Phenotypes Reveals Novel MKS Alleles and Uncovers a Specific Genetic Interaction between osm-3 and nphp-4.
PLoS genetics[HIGH INCIDENCE AND BROAD GENETIC VARIABILITY OF MECKEL-GRUBER SYNDROME IN THE ARAB POPULATION RESIDING IN NORTH-EAST ISRAEL].
HarefuahEnhanced diagnostic yield in Meckel-Gruber and Joubert syndrome through exome sequencing supplemented with split-read mapping.
BMC medical geneticsConserved Genetic Interactions between Ciliopathy Complexes Cooperatively Support Ciliogenesis and Ciliary Signaling.
PLoS geneticsPrenatal Diagnosis of Walker-Warburg Syndrome Using Single Nucleotide Polymorphism Array: A Clinical Experience from Three Related Palestinian Families with Congenital Hydrocephalus.
AJP reportsThe Ciliopathy Protein CC2D2A Associates with NINL and Functions in RAB8-MICAL3-Regulated Vesicle Trafficking.
PLoS geneticsFormation of the transition zone by Mks5/Rpgrip1L establishes a ciliary zone of exclusion (CIZE) that compartmentalises ciliary signalling proteins and controls PIP2 ciliary abundance.
The EMBO journalA review of Meckel-Gruber syndrome--incidence and outcome in the state of Qatar.
The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstetricians[Polydactyly, holoprosencephaly, cleft lip and cleft palate are not always what they seem: Case report].
Archivos argentinos de pediatriaHepatorenal fibrocystic diseases in children.
Pediatric nephrology (Berlin, Germany)A missense mutation in TMEM67 causes Meckel-Gruber syndrome type 3 (MKS3): a family from China.
International journal of clinical and experimental pathologyIdentification of a novel MKS locus defined by TMEM107 mutation.
Human molecular geneticsNDRC: A Disease-Causing Genes Prioritized Method Based on Network Diffusion and Rank Concordance.
IEEE transactions on nanobioscienceThe role of primary cilia in corpus callosum formation is mediated by production of the Gli3 repressor.
Human molecular geneticsMeckel Gruber syndrome, A case report.
OrganogenesisThe Meckel-Gruber syndrome protein TMEM67 controls basal body positioning and epithelial branching morphogenesis in mice via the non-canonical Wnt pathway.
Disease models & mechanismsSyndromic ciliopathies: From single gene to multi gene analysis by SNP arrays and next generation sequencing.
Molecular and cellular probesMeckel-Gruber Syndrome with unilateral renal agenesis.
Journal of the College of Physicians and Surgeons--Pakistan : JCPSPTMEM231, mutated in orofaciodigital and Meckel syndromes, organizes the ciliary transition zone.
The Journal of cell biologyGenetic, chromosomal, and syndromic causes of neural tube defects.
Saudi medical journalAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Meckel.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Meckel
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Missense Variants in the Second Transmembrane Domain of TMEM17 Disrupt Its Stability and Function and Lead to a Wide Phenotypic Spectrum of Ciliopathies.
- CRISPR-Cas9-Generated TXNDC15 c.560delA Homozygous Mouse Model Exhibits Meckel-Gruber Syndrome Phenotype.
- Ciliopathy-related B9 protein complex regulates ciliary axonemal microtubule posttranslational modifications and initiation of ciliogenesis.
- Biallelic Variants in TMEM17 Cause Meckel-Gruber Syndrome Within the Ciliopathy Spectrum.
- Cleavage of the Meckel-Gruber syndrome protein TMEM67 by ADAMTS9 uncouples Wnt signaling and ciliogenesis.
- Novel Compound Heterozygous Variants in the TCTN2 Gene Causing Meckel-Gruber Syndrome 8 in a Non-Consanguineous Chinese Family.
- Compound heterozygous missense and intronic variants in B9D1 contribute to a recurrent Meckel syndrome pedigree.
- First-trimester diagnosis of Meckel syndrome by ultrasonography with suspected mutation of CC2D2A: a case description.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:564(Orphanet)
- MONDO:0018921(MONDO)
- GARD:3436(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q1915681(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
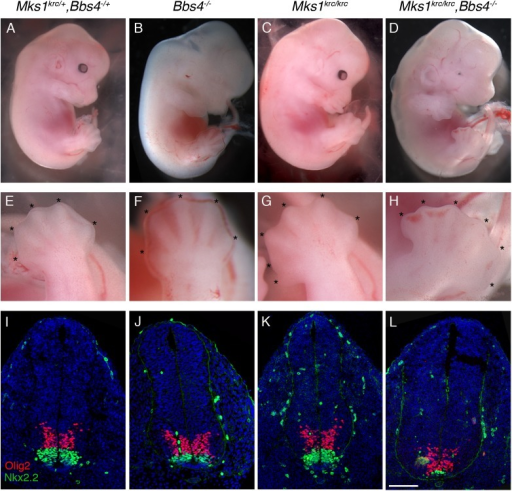
Síndrome Meckel
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov