Um caso de doença renal cística causada por uma modificação herdada do genoma do indivíduo.
Introdução
O que você precisa saber de cara
Um caso de doença renal cística causada por uma modificação herdada do genoma do indivíduo.
Tem tratamento?
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 41 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 128 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
14 genes identificados com associação a esta condição.
Curadoria gene-doença
fontes oficiaisAs a co-chaperone for HSPA5 it is required for proper folding, trafficking or degradation of proteins (PubMed:10827079, PubMed:15525676, PubMed:29706351). Binds directly to both unfolded proteins that are substrates for ERAD and nascent unfolded peptide chains, but dissociates from the HSPA5-unfolded protein complex before folding is completed (PubMed:15525676). May help recruiting HSPA5 and other chaperones to the substrate. Stimulates HSPA5 ATPase activity (PubMed:10827079). It is necessary fo
Endoplasmic reticulum lumen
Polycystic kidney disease 6 with or without polycystic liver disease
A form of polycystic kidney disease, a disorder characterized by progressive formation and enlargement of cysts in both kidneys, typically leading to end-stage renal disease in adult life. Cysts also occur in other organs, particularly the liver. PKD6 inheritance is autosomal dominant.
Mannosyltransferase that operates in the biosynthetic pathway of dolichol-linked oligosaccharides, the glycan precursors employed in protein asparagine (N)-glycosylation. The assembly of dolichol-linked oligosaccharides begins on the cytosolic side of the endoplasmic reticulum membrane and finishes in its lumen. The sequential addition of sugars to dolichol pyrophosphate produces dolichol-linked oligosaccharides containing fourteen sugars, including two GlcNAcs, nine mannoses and three glucoses.
Endoplasmic reticulum membrane
Component of the IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs) (PubMed:20889716, PubMed:22503633). Plays a pivotal role in proper development and function of ciliated cells through its role in ciliogenesis and/or cilium maintenance (PubMed:22503633). Required for the development and maintenance of the outer segments of rod and cone photoreceptor cells. Plays a role in maintenance and the delivery of opsin to
Cytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCell projection, cilium
Short-rib thoracic dysplasia 9 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome. SRTD9 is characterized by phalangeal cone-shaped epiphyses, chronic renal disease, nearly constant retinal dystrophy, and mild radiographic abnormality of the proximal femur. Occasional features include short stature, cerebellar ataxia, and hepatic fibrosis.
Putative RNA-binding protein. Acts as a negative regulator of Wnt signaling. May be involved in regulating gene expression during embryonic development
Cytoplasm
Renal dysplasia, cystic
An anomaly of the kidney characterized by numerous renal cysts and apparent disorder of differentiation of the renal parenchyma. Kidney of affected individuals lack the normal renal bean shape, and the collection drainage system. The cystic, dysplastic kidney contains undifferentiated mesenchyme, cartilaginous tissue, and immature collecting ducts.
Catalytic component of the TSC-TBC complex, a multiprotein complex that acts as a negative regulator of the canonical mTORC1 complex, an evolutionarily conserved central nutrient sensor that stimulates anabolic reactions and macromolecule biosynthesis to promote cellular biomass generation and growth (PubMed:12172553, PubMed:12271141, PubMed:12842888, PubMed:12906785, PubMed:15340059, PubMed:22819219, PubMed:24529379, PubMed:28215400, PubMed:33436626, PubMed:35772404). Within the TSC-TBC complex
Lysosome membraneCytoplasm, cytosol
Tuberous sclerosis 2
An autosomal dominant multi-system disorder that affects especially the brain, kidneys, heart, and skin. It is characterized by hamartomas (benign overgrowths predominantly of a cell or tissue type that occurs normally in the organ) and hamartias (developmental abnormalities of tissue combination). Clinical manifestations include epilepsy, learning difficulties, behavioral problems, and skin lesions. Seizures can be intractable and premature death can occur from a variety of disease-associated causes.
Functions in biogenesis and organization of the apical membrane of epithelial cells of the thick ascending limb of Henle's loop (TALH), where it promotes formation of complex filamentous gel-like structure that may play a role in the water barrier permeability (Probable). May serve as a receptor for binding and endocytosis of cytokines (IL-1, IL-2) and TNF (PubMed:3498215). Facilitates neutrophil migration across renal epithelia (PubMed:20798515) In the urine, may contribute to colloid osmotic p
Apical cell membraneBasolateral cell membraneCell projection, cilium membraneSecreted
Tubulointerstitial kidney disease, autosomal dominant 1
A form of autosomal dominant tubulointerstitial kidney disease, a genetically heterogeneous disorder characterized by slowly progressive loss of kidney function, bland urinary sediment, hyperuricemia, absent or mildly increased albuminuria, lack of severe hypertension during the early stages, and normal or small kidneys on ultrasound. Renal histology shows variable abnormalities including interstitial fibrosis with tubular atrophy, microcystic dilatation of the tubules, thickening of tubular basement membranes, medullary cysts, and secondary glomerulosclerotic or glomerulocystic changes with abnormal glomerular tufting. There is significant variability, as well as incomplete penetrance.
Required for renal tubular integrity. May regulate local cytoskeletal structure in kidney tubule epithelial cells. May regulate ciliary biogenesis through targeting of proteins to the cilia (PubMed:37598857). Plays a role in organogenesis, and is involved in the regulation of the Hippo signaling pathway (PubMed:26967905)
CytoplasmCytoplasm, cytoskeletonCell projection, ciliumCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, cilium axoneme
Nephronophthisis 9
An autosomal recessive disorder resulting in end-stage renal disease. It is a progressive tubulo-interstitial kidney disorder histologically characterized by modifications of the tubules with thickening of the basement membrane, interstitial fibrosis and, in the advanced stages, medullary cysts.
Dolichyl-phosphate beta-glucosyltransferase that operates in the biosynthetic pathway of dolichol-linked oligosaccharides, the glycan precursors employed in protein asparagine (N)-glycosylation. The assembly of dolichol-linked oligosaccharides begins on the cytosolic side of the endoplasmic reticulum membrane and finishes in its lumen. The sequential addition of sugars to dolichol pyrophosphate produces dolichol-linked oligosaccharides containing fourteen sugars, including two GlcNAcs, nine mann
Endoplasmic reticulum membrane
Polycystic kidney disease 7
A form of polycystic kidney disease, a disorder characterized by progressive formation and enlargement of cysts in both kidneys, typically leading to end-stage renal disease in adult life. Cysts also occur in other organs, particularly the liver. PKD7 inheritance is autosomal dominant.
Involved in primary cilium formation (PubMed:19852954, PubMed:28530676). Probably acts as a transition zone protein required for localization of PKD1/PC1 and PKD2/PC2 to the ciliary membrane (PubMed:28530676)
Cytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriole
Polycystic kidney disease 5
A form of polycystic kidney disease, a disorder characterized by progressive formation and enlargement of cysts in both kidneys, typically leading to end-stage renal disease in adult life. Cysts may also occur in other organs, particularly the liver. PKD5 inheritance is autosomal recessive.
Promotes ciliogenesis in renal epithelial cells and therefore participates in the tubules formation and/ or ensures the maintenance of the architecture of the lumen of the kidney (By similarity). Has an impact on cellular symmetry by ensuring correct bipolar cell division through the regulation of centrosome duplication and mitotic spindle assembly and by maintaining oriented cell division (OCD) during tubular elongation through planar cell polarity (PCP) pathway (PubMed:20554582). During epithe
Cell membraneCytoplasmCell projection, ciliumCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, spindleChromosome, centromereApical cell membraneNucleusSecreted, extracellular exosomeSecretedEndoplasmic reticulumGolgi apparatus
Polycystic kidney disease 4, with or without polycystic liver disease
A severe form of polycystic kidney disease affecting the kidneys and, in some cases, the hepatic biliary tract. The clinical spectrum is widely variable, with most cases presenting during infancy. The fetal phenotypic features classically include enlarged and echogenic kidneys, as well as oligohydramnios secondary to a poor urine output. Up to 50% of the affected neonates die shortly after birth, as a result of severe pulmonary hypoplasia and secondary respiratory insufficiency. In the subset that survives the perinatal period, morbidity and mortality are mainly related to severe systemic hypertension, renal insufficiency, and portal hypertension due to portal-tract fibrosis. PKD4 inheritance is autosomal recessive.
Catalytic subunit of glucosidase II that cleaves sequentially the 2 innermost alpha-1,3-linked glucose residues from the Glc(2)Man(9)GlcNAc(2) oligosaccharide precursor of immature glycoproteins (PubMed:10929008). Required for PKD1/Polycystin-1 and PKD2/Polycystin-2 maturation and localization to the cell surface and cilia (PubMed:27259053)
Endoplasmic reticulumGolgi apparatusMelanosome
Polycystic kidney disease 3 with or without polycystic liver disease
A form of polycystic kidney disease, a disorder characterized by progressive formation and enlargement of cysts in both kidneys, typically leading to end-stage renal disease in adult life. Cysts also occur in other organs, particularly the liver. PKD3 inheritance is autosomal dominant.
The alpha subunit has cell adhesive properties. Can act both as an adhesion and an anti-adhesion protein. May provide a protective layer on epithelial cells against bacterial and enzyme attack The beta subunit contains a C-terminal domain which is involved in cell signaling, through phosphorylations and protein-protein interactions. Modulates signaling in ERK, SRC and NF-kappa-B pathways. In activated T-cells, influences directly or indirectly the Ras/MAPK pathway. Promotes tumor progression. Re
Apical cell membraneSecretedCell membraneCytoplasmNucleus
Forms a nonselective cation channel (PubMed:11854751, PubMed:11991947, PubMed:15692563, PubMed:26269590, PubMed:27071085, PubMed:31441214, PubMed:39009345). Can function as a homotetrameric ion channel or can form heteromer with PKD1 (PubMed:31441214, PubMed:33164752). Displays distinct function depending on its subcellular localization and regulation by its binding partners (PubMed:11854751, PubMed:11991947, PubMed:27214281, PubMed:29899465). In primary cilium functions as a cation channel, wit
Cell projection, cilium membraneEndoplasmic reticulum membraneCell membraneBasolateral cell membraneCytoplasmic vesicle membraneGolgi apparatusVesicleSecreted, extracellular exosome
Polycystic kidney disease 2 with or without polycystic liver disease
An autosomal dominant disorder characterized by progressive formation and enlargement of cysts in both kidneys, typically leading to end-stage renal disease in adult life. Cysts also occurs in the liver and other organs. It represents approximately 15% of the cases of autosomal dominant polycystic kidney disease. PKD2 is clinically milder than PKD1 but it has a deleterious impact on overall life expectancy.
Component of a heteromeric calcium-permeable ion channel formed by PKD1 and PKD2 that is activated by interaction between PKD1 and a Wnt family member, such as WNT3A and WNT9B (PubMed:27214281). Both PKD1 and PKD2 are required for channel activity (PubMed:27214281). Involved in renal tubulogenesis (PubMed:12482949). Involved in fluid-flow mechanosensation by the primary cilium in renal epithelium (By similarity). Acts as a regulator of cilium length, together with PKD2 (By similarity). The dynam
Cell membraneCell projection, ciliumEndoplasmic reticulumGolgi apparatusVesicleSecreted, extracellular exosome
Polycystic kidney disease 1 with or without polycystic liver disease
An autosomal dominant disorder characterized by renal cysts, liver cysts and intracranial aneurysm. Clinical variability is due to differences in the rate of loss of glomerular filtration, the age of reaching end-stage renal disease and the occurrence of hypertension, symptomatic extrarenal cysts, and subarachnoid hemorrhage from intracranial 'berry' aneurysm.
Medicamentos e terapias
Mecanismo: Vasopressin V2 receptor antagonist
Mecanismo: Type-1 angiotensin II receptor antagonist
Mecanismo: Angiotensin-converting enzyme inhibitor
Mecanismo: Somatostatin receptor 5 agonist
Mecanismo: Mitochondrial complex I (NADH dehydrogenase) inhibitor
Variantes genéticas (ClinVar)
532 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
34 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Doença renal cística genética
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
2 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Reversible cystogenesis in juvenile primate ADPKD models: evidence from PKD1 heterozygous monkeys.
Autosomal dominant polycystic kidney disease (ADPKD) is a common inherited disorder caused predominantly by heterozygous mutations in the PKD1 gene, leading to progressive renal cyst formation. While PKD1 mutant mouse models have provided mechanistic insights, PKD1 heterozygous mice fail to replicate the early cystogenesis observed in human patients. To address this gap, we conducted a longitudinal study using PKD1 heterozygous cynomolgus monkeys. Serial renal ultrasonography from birth to five years of age-corresponding to human childhood-revealed progressive cyst development. Remarkably, a subset of cysts, particularly smaller ones, exhibited spontaneous regression over time. This phenomenon was also observed in PKD1 mosaic monkeys harboring mixed variant patterns. In contrast, monkeys with biallelic PKD1 loss-of-function variants developed severe cystic disease, kidney enlargement, hepatic cysts, and elevated serum creatinine, resembling the clinical features of advanced ADPKD. These findings demonstrate that early renal cysts may possess intrinsic plasticity, challenging the conventional view of ADPKD as a relentlessly progressive disorder. Our results suggest that the early stages of cystogenesis represent a potential therapeutic window for intervention, in which cyst regression may be promoted. The PKD1 heterozygous monkey model thus provides a valuable platform for studying the developmental dynamics of ADPKD and for evaluating novel therapeutic approaches targeting early cystic changes.
Heterogeneous regulation of fibroblast growth factor 23 in acute kidney injury, chronic kidney disease, and polycystic kidney disease: mechanisms, diagnostic utility, and clinical implications.
Fibroblast Growth Factor 23 (FGF23) is a bone-derived hormone regulating phosphate and vitamin D metabolism, now recognized as a dynamic biomarker across acute and chronic kidney disorders. Elevated FGF23 is a hallmark of chronic kidney disease (CKD), but also rises acutely in acute kidney injury (AKI) and appears disproportionately high in autosomal dominant polycystic kidney disease (ADPKD), underscoring condition-specific regulation. This review explores the correlation and heterogeneity of FGF23 expression in AKI, CKD, and ADPKD, highlighting shared and divergent mechanisms and the diagnostic and therapeutic implications. We summarize FGF23 expression kinetics in each condition, elucidate known and proposed molecular drivers of its elevation, and discuss how FGF23 serves as a unifying yet disease-divergent marker in renal pathology. In AKI, inflammation, ischemia, and acute metabolic stress drive a rapid FGF23 surge, whereas in CKD, phosphate retention and Klotho deficiency promote a sustained, maladaptive FGF23 elevation. ADPKD shows early FGF23 increases independent of glomerular filtration rate (GFR), potentially due to ectopic production (liver and cysts) and unique tubular defects. Clinically, FGF23 has emerged as an indicator of disease severity and outcomes in these contexts: it can signal early AKI and predict progression, is a strong prognostic factor for mortality and cardiovascular complications in CKD, and correlates with cystic disease burden and kidney growth in ADPKD. We also examine FGF23's systemic effects (notably on cardiovascular remodeling) and potential therapeutic targets, from modulating phosphate balance and iron metabolism to novel interventions in development. Understanding the nuanced regulation of FGF23 across acute injury, chronic degeneration, and genetic kidney disease provides insight into acute-chronic disease intersections and guides precision diagnostics and therapies for improved patient outcomes.
Emodin retards renal cyst progression in ADPKD by inhibiting cell proliferation and decreasing oxidative stress.
Autosomal dominant polycystic kidney disease (ADPKD) is a common monogenic genetic disease characterized by bilateral renal cyst formation and enlargement. The purpose of this study was to explore the therapeutic effects of emodin, which is a key active constituent of traditional Chinese herbal medicine Rhubarb, on ADPKD and its underlying mechanisms. Herein, we found emodin significantly inhibited renal cyst progression in ADPKD. RNA-sequencing analysis and network pharmacological analysis indicated that emodin may exert its therapeutic effects by modulating cell proliferation and oxidative stress, in which the Wnt and Nrf2 related signaling pathways probably played important roles. These findings were subsequently verified. Emodin was shown to downregulate cell proliferation and oxidative stress levels, inhibit the proliferation-associated Wnt/β-catenin signaling pathway, and activate the antioxidant Nrf2/GPX4 signaling pathway. Furthermore, we found that the Wnt pathway exerted an inhibitory effect on Nrf2/GPX4 axis upstream. Through inhibition of the Wnt pathway, emodin may relieve its suppression of Nrf2/GPX4 axis, ultimately achieving dual modulation of proliferation and oxidative stress. RNA-sequencing analysis also indicated that Wnt7a was the most significantly altered Wnt pathway-related gene following emodin treatment in ADPKD. We confirmed that emodin reduced both the mRNA and protein levels of Wnt7a. Emodin also potentially acted as a direct binder to Wnt7a, this might be the upstream mechanism by which it modulated the Wnt pathway. Overall, our study suggested that emodin may serve as a potential therapeutic compound for ADPKD treatment.
An NGS-based investigation of copy number variants in the diagnosis and severity of adult polycystic kidney disease.
Polycystic kidney disease (PKD) is a Mendelian renal disease characterised by the development of cysts and progressive decline in kidney function, leading to kidney failure. Although genetic testing can provide a precise molecular diagnosis of PKD in the majority of cases, 6-13% of patients remain unsolved. Copy number variants (CNVs) are an established pathogenic mechanism in PKD, however detection typically relies on multiplex ligation-dependent probe amplification (MLPA) which is resource intensive and separate to next-generation sequencing (NGS) pipelines. Here, a bioinformatics tool ClinCNV was used to call CNVs from NGS data of 371 people with PKD who had previously undergone short nucleotide variant (SNV) analysis with a standard NGS pipeline. Diagnostic CNVs were confirmed in 13 patients across 7 families, increasing the diagnostic yield from 86.5 to 90.0%. We also tested CNVs as potential disease modifiers. Regression models indicated an association of cystic gene duplication burden to worse kidney survival (HR = 1.56, 95% CI: 1.26, 1.93, adj-p = 0.0004). These models also revealed that duplication burden in genes unrelated to cystic kidney disease associated with the absence of liver cysts, possibly driven by a region containing LRP5L. These results demonstrate the utility of targeted gene panel and exome sequencing for the detection of CNVs in key PKD genes.
Epithelial Dynamics of Cystogenesis in Genetic Models of Autosomal Dominant Polycystic Kidney Disease.
Autosomal dominant polycystic kidney disease (ADPKD), caused by mutations in PKD1 or PKD2, is characterized by progressive and exponential enlargement of renal and hepatic cysts. However, the epithelial dynamics that generate this growth pattern remain incompletely understood. Using Brainbow/Confetti multicolor clonal lineage tracing in developmental and adult-onset ADPKD mouse models, we show that polycystin-deficient epithelial cells initiate clonal expansion at early stages of tubule dilation and continue to expand throughout cyst progression. Concurrently, cyst-lining cells undergo a progressive transition from columnar to flattened morphology, which amplifies luminal enlargement independent of cell number. Integrating these measures, we developed a mathematical model demonstrating that the combination of this clonal expansion and epithelial cell shape remodeling is sufficient to produce the exponential growth trajectory observed in ADPKD. Together, these findings define the core epithelial mechanisms that drive cyst initiation and expansion, and may provide a mathematical framework for the emergent exponential growth of cysts.
Publicações recentes
Cystic renal diseases: role of ultrasound. Part II, genetic cystic renal diseases.
Cystic renal diseases: role of ultrasound. Part I, non-genetic cystic renal diseases.
📚 EuropePMCmostrando 198
Rapidly Progressive Kidney Failure With Transient Non-Cystic Kidney Enlargement: A Case Report Highlighting Delayed Medullary Cyst Formation.
Nephrology (Carlton, Vic.)[Genetic kidney diseases].
Deutsche medizinische Wochenschrift (1946)ALDH1A1 is a potential novel target for treatment of ADPKD.
Kidney internationalMeckel-Gruber syndrome: a rare and fatal congenital disorder (case report).
The Pan African medical journalGlycolytic alterations as biomarkers in polycystic kidney disease: A study using a PKD1 knockout model in NRK-52E rat kidney epithelial cells.
Physiological reportsReversible cystogenesis in juvenile primate ADPKD models: evidence from PKD1 heterozygous monkeys.
Human molecular geneticsMachine Learning Prediction of Progression to Dialysis in Patients With Polycystic Kidney Disease: Population-Based Retrospective Cohort Study.
JMIR medical informatics[Autosomal dominant polycystic kidney disease].
Revue medicale de LiegeGGPS1 Promoter Variant (rs3806394) Is Associated With Larger Simple Renal Cysts via Reduced GGPPS Expression.
Human mutationExome sequencing in patients with medullary sponge kidney.
Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal AssociationHeterogeneous regulation of fibroblast growth factor 23 in acute kidney injury, chronic kidney disease, and polycystic kidney disease: mechanisms, diagnostic utility, and clinical implications.
Frontiers in molecular biosciencesALG8-Driven Metabolic Reprogramming in Polycystic Kidney Disease: A Systematic Synthesis of Evidence Linking Glycosylation Defects to Metabolic Signaling.
IUBMB lifeUnilateral Multicystic Dysplastic Kidney in a Fetus Associated With Parental Genetic and Environmental Risk Factors: A Case Report.
CureusMultifactorial Predictors of Renal Outcomes in Alport Syndrome: Integrating Genetic, Clinical, and Cystic Phenotypes.
Kidney medicineInflammation-Driven Field Cancerization in End-Stage Kidney Disease.
Cancer discoveryMulticystic Kidney Disease in a Family With Tuberous Sclerosis Complex.
Nephrology (Carlton, Vic.)The Inheritance Puzzle: A Case of Dual Genetic Kidney Disease.
Nephrology (Carlton, Vic.)Next-Generation Sequencing Defines a Molecularly Confirmed ARPKD Core Within the Broader PKHD1-Associated Disease Spectrum.
GenesBilateral Glomerulocystic Kidney Disease With Extensive Embryonal Hyperplasia in a Setting of HNF1B Mutation.
Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology SocietyEmodin retards renal cyst progression in ADPKD by inhibiting cell proliferation and decreasing oxidative stress.
Life sciencesSevere Renal Phenotype Across A Multigenerational Tuberous Sclerosis Complex (TSC) Family.
Molecular genetics & genomic medicineAn NGS-based investigation of copy number variants in the diagnosis and severity of adult polycystic kidney disease.
European journal of human genetics : EJHGELOC-mutated Renal Cell Carcinoma: Clinicopathologic, Immunohistochemical, and Molecular Genetic Analysis of 35 Cases.
Modern pathology : an official journal of the United States and Canadian Academy of Pathology, IncGLIS3, a novel regulator of eicosanoid gene expression and metabolism in normal kidney and polycystic kidney disease.
Biochemical pharmacologyBICC1 interacts with PKD1 and PKD2 to drive cystogenesis in ADPKD.
eLifeEpithelial Dynamics of Cystogenesis in Genetic Models of Autosomal Dominant Polycystic Kidney Disease.
CellsA HaloTag Knock-In Resource for In Vivo and In Vitro Analysis of Endogenous Polycystin-2 Localization, Turnover, and Transport.
Journal of the American Society of Nephrology : JASNDual Monogenic Cystic Disease Case Report: Autosomal Dominant Polycystic Kidney Disease and Autosomal Dominant Polycystic Liver Disease.
Clinical case reportsInvestigation of Urinary Extracellular Vesicles as Novel and Safe Therapeutics for Autosomal Recessive Polycystic Kidney Disease (ARPKD).
Journal of biomedical materials research. Part A[Clinicopathological and molecular features of acquired cystic disease-associated renal cell carcinoma].
Zhonghua bing li xue za zhi = Chinese journal of pathologyVascular transcriptional and metabolic changes precede progressive intrarenal microvascular rarefaction in autosomal dominant polycystic kidney disease.
American journal of physiology. Renal physiology[A Family case of von Hippel-Lindau syndrome].
Problemy endokrinologiiIdentification of Pathogenic PKHD1 Variants in Infants with Autosomal Recessive Polycystic Kidney Disease from the Dhofar Region, Oman.
F1000ResearchMorphometric and Functional Evaluation of Nonrenal Vasculature in Female and Male PKD/Mhm (Cy/+) Rats.
Kidney & blood pressure researchA rare early-onset bilateral renal cysts, focal seizures in a 1-year-old male with tuberous sclerosis and No mutation identified.
Oxford medical case reportsZebrafish as a Model System for Polycystic Kidney Disease: Lessons from ift140 Mutants.
Journal of the American Society of Nephrology : JASNThe Role of Immune Cells as Modulators of Progression in Polycystic Kidney Disease: A Systematic Review.
Kidney360Integrin α 1 β 1 Promotes Interstitial Fibrosis and Cyst Growth in a Mouse Model of Polycystic Kidney Disease.
Journal of the American Society of Nephrology : JASNDisruption of a six-nucleotide miRNA motif improves PKD1 dosage and ameliorates polycystic kidney disease.
Nucleic acids researchCoincidence of autosomal dominant polycystic kidney disease and Alport syndrome: a case report and literature review.
CEN case reportsPrevalence and Impact of Chronic Liver Disease in Adult Patients Admitted With Cystic Fibrosis.
CureusCRISPR-Cas9-Generated TXNDC15 c.560delA Homozygous Mouse Model Exhibits Meckel-Gruber Syndrome Phenotype.
Genesis (New York, N.Y. : 2000)Distinct genomic, microenvironmental, and nephron signatures in VHL kidney cysts and tumors.
Scientific reportsGeneration of Mice Harboring Bicc1 Conditional Null Alleles.
Genesis (New York, N.Y. : 2000)Loss of Snhg5 disrupts cell-cycle regulation without altering cystogenesis in a mouse model of polycystic kidney disease.
Scientific reportsPAX gene expression in autosomal dominant polycystic kidney disease contributes to cyst expansion and regulates a gene network associated with cyst growth.
Human molecular geneticsTargeted Next-Generation Sequencing of MEN 1, RET, CDC 73, and CDKNIB Genes in Familial Primary Hyperparathyroidism: A Study from Northern India.
Indian journal of endocrinology and metabolismGenetic Testing in Cystic Kidney Disease.
Kidney360Lack of ANKMY2 suppresses kidney cystogenesis in embryonic- and adult-onset polycystic kidney disease.
PLoS geneticsMulti-Target Effects of Novel Synthetic Indanedione-Based Spirotransdicalinol Compound 4l in Autosomal Dominant Polycystic Kidney Disease.
FASEB journal : official publication of the Federation of American Societies for Experimental BiologyPrenatal Diagnosis and Postnatal Outcomes of Fetal ADPKD: A Single-Center Retrospective Cohort Study.
Medicina (Kaunas, Lithuania)Clinical characteristics and genotype-phenotype correlation for patients with autosomal recessive polycystic kidney disease and PKHD1 mutations.
Clinical nephrologyFunctional Coupling of Calcium-Sensing Receptor and Polycystin-2 in Renal Epithelial Cells: Physiological Role and Potential Therapeutic Target in Polycystic Kidney Disease.
International journal of molecular sciencesComplexities of Bartter Syndrome Type III: A Case Study in Jordan.
Oxford medical case reportsCase Report: Neonatal nephropathy with polycystic appearance in child harboring WT1 variant.
Frontiers in pediatrics[Monogenic Kidney Diseases in Children - Five Years of Experience from a Dedicated Nephrogenetic Clinic in Southern Israel].
HarefuahBirt-Hogg-Dubé Syndrome: A Mini Review of the Clinical Manifestations, Investigation, and Management.
Journal of personalized medicineFrom mutation to symptoms: a multi-center study on HNF1B-related nephropathy in Chinese children.
BMC nephrologyOncogenic NRAS Mutation in Incipient Sarcomatous Transformation of Cystic Nephroma From a Patient With DICER1-Related Tumor Predisposition Syndrome.
Pediatric blood & cancerPredictors of kidney survival in children with autosomal recessive polycystic kidney disease.
Clinical and experimental nephrologyAnticodon-edited tRNA enables translational readthrough of COL4A5 premature termination codons.
PloS onePrenatal diagnosis to postnatal outcomes in multicystic dysplastic kidney: experience of a tertiary center in the Black Sea region.
Revista da Associacao Medica Brasileira (1992)PERADIGM: Phenotype embedding similarity-based rare disease gene mapping.
PLoS geneticsAldehyde dehydrogenase 1 A1 regulates the transcription of PD-L1 and its targeting with disulfiram together with PD-L1 immunotherapy synergistically delays cyst growth in ADPKD.
Kidney internationalPhenotypic and genotypic analysis of pediatric nephronophthisis patients with different levels of proteinuria.
Renal failureIn vivo base editing rescues ADPKD in a humanized mouse model.
Nature communicationsZONAB regulates renal cyst formation in nphp1 knockout mice.
Translational research : the journal of laboratory and clinical medicineExome Sequencing in a Large Cohort with Ciliopathy-Related Kidney Disease.
Clinical journal of the American Society of Nephrology : CJASNSegmentation of renal tubules and automatic biomarker quantification in ciliopathy preclinical models.
Annual International Conference of the IEEE Engineering in Medicine and Biology Society. IEEE Engineering in Medicine and Biology Society. Annual International ConferenceCiliary ARLs drive renal cystogenesis.
bioRxiv : the preprint server for biologyPrenatal-Onset Autosomal Dominant Polycystic Kidney Disease: Clinical Spectrum and Genetic Complexity of a Pseudo-Recessive Phenotype.
Prenatal diagnosisNovel Compound Heterozygous Variants in the TCTN2 Gene Causing Meckel-Gruber Syndrome 8 in a Non-Consanguineous Chinese Family.
Molecular genetics & genomic medicineSenior-Løken syndrome with IQCB1/NPHP5 mutation in an adult: a case report.
Journal of medical case reportsExploring the Role of Plant-Based Nutrition in Polycystic Kidney Disease.
NutrientsNon-Coding RNAs as Emerging Regulators in Kidney Pathophysiology: From Molecular Mechanisms to Therapeutic Potential.
GenesAtypical Liver Ultrasound Image in a Boy with Autosomal Recessive Polycystic Kidney Disease (ARPKD) and New PKD1 Variant-A Case Report.
GenesCoronary aneurysms and dissections in a patient with autosomal dominant polycystic kidney disease: a case report.
Journal of medical case reportsPresentation of Patients With Congenital Anomalies of the Kidney and Urinary Tract and PAX2 Loss-of-Function Variants and Implications for Clinical Management.
Kidney international reportsGenotype-first assessment of presentation and penetrance of neurofibromatosis type 1, autosomal dominant polycystic kidney disease, and Marfan syndrome within the All of Us research program cohort.
Genetics in medicine : official journal of the American College of Medical GeneticsTranslational readthrough therapy for ADPKD induces polycystin1 expression and partially rescues functional deficits in PKD1 mutant cells.
Scientific reportsJoubert syndrome 26 protein enforces compartmentalized motility of a ciliary kinesin.
Proceedings of the National Academy of Sciences of the United States of AmericaSpatial Multiomic Analyses Reveal Carcinogenic Pathways in End-Stage Renal Disease.
Cancer discoveryIn-depth 3D exploration of autosomal dominant polycystic kidney disease through light sheet fluorescence microscopy.
Scientific reportsDe novo PKD1 splicing and missense variants in two familial ADPKD: Molecular characterization and genetic counseling implications.
GeneTheratyping as a tool to guide clinical decision-making in patients with suspected cystic fibrosis and variants currently ineligible for CFTR modulator therapy.
Respiratory medicineComprehensive dietary patterns explain acquired cystic kidney disease risk through genetic and metabolomic mechanisms.
Frontiers in nutritionInhibition of the inflammasome ameliorates orthologous polycystic kidney disease.
Proceedings of the National Academy of Sciences of the United States of AmericaNovel PIBF1 Pathogenic Variant in Three Siblings with Joubert Syndrome Type 33.
Molecular syndromologyAtypical presentations of fetal polycystic kidney disease demonstrates the utility of a genomic autopsy for accurate post-mortem diagnoses.
Human genomicsA TRIM8 Variant in a Child: Neuro-Renal Syndrome Causing Features Suggestive of Medullary Sponge Kidney.
Nephrology (Carlton, Vic.)Molecular pathology and cystogenic propensity of the ADPKD Taiwan founder variant.
JCI insightA Case of TSC2/PKD1 Contiguous Gene Deletion Syndrome With Proven Kidney Pathology.
Kidney medicineSupportive care in life-limiting, non-malignant chronic disease: scoping review.
BMJ supportive & palliative careCulture and Adaptation of Pseudomonas aeruginosa During Early Infection in Children With Cystic Fibrosis.
The Journal of infectious diseasesEliosin-an alternative product from the HmPKD1 locus is a component of endoplasmic reticulum mitochondria membrane contact sites.
PloS oneCiliopathy-related B9 protein complex regulates ciliary axonemal microtubule posttranslational modifications and initiation of ciliogenesis.
The Journal of clinical investigationUnraveling the Genetic Links Between Polycystic Kidney Disease and Hypertension Through ARL13B.
International journal of nephrology and renovascular diseaseHypertension Resistant to RAAS Inhibitors as a Prognostic Indicator for Rapid Progression to ESRD in ADPKD: A Ten-Year Follow-Up.
Diagnostics (Basel, Switzerland)Prenatal Diagnosis of Joubert Syndrome 23 With Left Isomerism: A Novel Phenotype Associated With Pathogenic KIAA0586 Variant.
Prenatal diagnosisElucidating Mechanisms of Hypomorphic WDR19-Related Kidney Failure.
Kidney international reportsExome Sequencing in Saudi Arabian Pediatric Kidney Disease Single-Center Cohort.
Kidney international reportsTackling ciliary specialization to understand phenotypic variability in human primary ciliopathies.
Journal of cell scienceContemporary management of advanced chronic kidney disease: An evidence-based review.
European journal of internal medicineKcnn4/KCa3.1 inhibition blunts polycystic kidney disease progression in mouse models.
JCI insightMED15-TFE3 rearranged renal cell carcinoma: a subtype of TFE3-rearranged renal cell carcinoma with unique clinicopathologic features and better prognosis.
World journal of surgical oncologyThe coming of age for polycystic kidney disease.
Kidney internationalMulticystic dysplastic kidneys (MCDK) during prenatal life and postnatal outcome.
Archives of gynecology and obstetricsInhibiting mPGES-2 impedes renal cyst growth in mice with polycystic kidney disease.
Acta pharmacologica SinicaGlobal burden of 292 causes of death in 204 countries and territories and 660 subnational locations, 1990-2023: a systematic analysis for the Global Burden of Disease Study 2023.
Lancet (London, England)Global age-sex-specific all-cause mortality and life expectancy estimates for 204 countries and territories and 660 subnational locations, 1950-2023: a demographic analysis for the Global Burden of Disease Study 2023.
Lancet (London, England)Burden of 375 diseases and injuries, risk-attributable burden of 88 risk factors, and healthy life expectancy in 204 countries and territories, including 660 subnational locations, 1990-2023: a systematic analysis for the Global Burden of Disease Study 2023.
Lancet (London, England)Interaction Proteomics of Polycystins 1 and 2 Reveal a Novel Role for the BLOC-1/BORC Lysosomal Positioning Complex.
Molecular & cellular proteomics : MCPPKD1 Splice and PKD2 Nonsense Variants Identified in Two Chinese Families With Autosomal Dominant Polycystic Kidney Disease.
Nephrology (Carlton, Vic.)Inactivation of Pkd2 in adult mice results in delayed cyst formation and identifies sex as a major modifier of disease severity.
Kidney internationalA tough job: ion channels, transporters, and pumps during organ fibrosis.
American journal of physiology. Cell physiologySpatial Transcriptomics Reveals Injured Cells, Signature Genes, and Communication Patterns in the Cyst Microenvironment of Polycystic Kidney Disease.
Journal of the American Society of Nephrology : JASNBiallelic Variants in TMEM17 Cause Meckel-Gruber Syndrome Within the Ciliopathy Spectrum.
Clinical geneticsZinc inhibits cAMP-induced Cl- secretion in intestinal epithelial cells via calcium-sensing receptor.
American journal of physiology. Cell physiologyEvaluating gene variations in autosomal dominant polycystic kidney disease patients using whole exome sequencing and phenotype to genotype analysis.
Renal failureCommemorating the National Institute of Diabetes and Digestive and Kidney Diseases' Advances in Kidney Health: 75 Years of Discovery and Impact.
Clinical journal of the American Society of Nephrology : CJASNGenome editing strategies to generate working models of polycystic kidney disease.
Scientific reportsAssociation between brain connectivity and renal pathophysiology: a multi-trait Mendelian randomization analysis.
Brain structure & functionPKD1 5'UTR variants are a rare cause of disease in ADPKD and suggest a new focus for therapeutic development.
European journal of human genetics : EJHGDifferential modulation of polycystin-2 gain-of-function channels by cysteine-reactive compounds, amphiphilic substances, and S4-S5 linker mutations.
The Journal of biological chemistryIdentification of PDIA6 Mutation in a Case of Autosomal Recessive Polycystic Kidney Disease: A Case Report and Review of Literature.
Clinical geneticsMonoallelic IFT140 Variants Causing Childhood-Onset Autosomal Dominant Polycystic Kidney Disease.
American journal of kidney diseases : the official journal of the National Kidney FoundationHyperfiltration and Elevated Fractional Excretion of Magnesium in Children with Cystic Fibrosis.
ACS pharmacology & translational scienceiPSC-based drug discovery identified the Hippo signaling pathway as a therapeutic target in the fibrosis of NPHP1-deficient nephronophthisis.
Stem cell research & therapyChronic kidney disease- mineral and bone disorder in autosomal dominant policystic kidney disease.
BoneCharacterization of complex renal cysts in hereditary leiomyomatosis and renal cell cancerUsing magnetic resonance based qualitative features.
Abdominal radiology (New York)Further evidence of RNU4ATAC variants causing Joubert syndrome with skeletal involvement.
Journal of medical geneticsA case of HDR syndrome with recurrent matured ovarian teratomas.
Endocrinology, diabetes & metabolism case reportsPATJ deficiency leads to cystic kidney disease and related ciliopathies.
HGG advancesDifferential regulation of NHE3 expression in type 1 and type 2 diabetic intestine: impaired endosomal regulation of NHE3 expression in type 1 diabetes.
American journal of physiology. Cell physiologyObesity and Kidney Disease: A Focus on Ciliopathies.
Giornale italiano di nefrologia : organo ufficiale della Societa italiana di nefrologiaResearch Progress on Risk Factors or Prediction Models for Ureteropelvic Junction Obstruction in Children.
Archivos espanoles de urologiaTimely excision of prophage Φ13 is essential for the Staphylococcus aureus infectious process.
Infection and immunityA CSPP1 variant associated with metabolic dysfunction in Joubert syndrome: a case report.
Journal of medical case reportsCleavage of N-terminus of polycystin-1 increases calcium permeability of polycystin-1/2 receptor channel complexes.
JCI insightBilateral, multicystic fumarate hydratase-deficient renal cell carcinoma in patient with hereditary leiomyomatosis & renal cell carcinoma syndrome: A case report and review of the literature.
Diagnostic pathologyTrifecta of Cysts: Unraveling the Diagnosis in a Patient With Renal, Hepatic, and Pancreatic Cysts.
CureusAutosomal Dominant Polycystic Kidney Disease: Core Curriculum 2025.
American journal of kidney diseases : the official journal of the National Kidney FoundationIn vivo base editing reduces liver cysts in autosomal dominant polycystic kidney disease.
Molecular therapy : the journal of the American Society of Gene TherapyMissense Variants in the Second Transmembrane Domain of TMEM17 Disrupt Its Stability and Function and Lead to a Wide Phenotypic Spectrum of Ciliopathies.
Clinical geneticsReversed cortico-medullary differentiation in kidneys on fetal magnetic resonance imaging - a case series.
Pediatric nephrology (Berlin, Germany)Systematic review of outcomes reported in clinical research on nephronophthisis: how do they align with SONG Kids priorities?
Pediatric nephrology (Berlin, Germany)Whole exome sequencing reveals a pathogenic homozygous CLDN16 mutation in a 17-year-old patient with familial hypomagnesemia with hypercalciuria and nephrocalcinosis: A case report.
MedicineThe impact of chronic kidney disease on arteriovenous fistula remodeling: studies in a murine model of autosomal dominant polycystic kidney disease.
American journal of physiology. Renal physiologyAssociation of tumor necrosis factor receptors 1 and 2 with kidney volume and function in patients with autosomal dominant polycystic kidney disease.
Clinical nephrologyIntronic and Coding Genetic Variants in Autosomal Recessive Polycystic Kidney Disease Among Israeli Bedouins of Arabian Peninsula Ancestry.
American journal of kidney diseases : the official journal of the National Kidney FoundationGLP-1RA Semaglutide Delays the Progression of ADPKD Through Regulation of Glycolysis, Mitochondria Function and Ketosis.
FASEB journal : official publication of the Federation of American Societies for Experimental BiologyRenal tissue-resident macrophages promote cystogenesis in early polycystic kidney disease.
Journal of cell scienceTransforming advancing autosomal dominant polycystic kidney disease care: investigating new horizons in treatment and research.
InflammopharmacologyKDIGO 2025 ADPKD guideline: a commentary on diagnosis and management of hepatopancreatic manifestations by the ERA Working Group Genes & Kidney.
Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal AssociationTypical and atypical ADPKD: predicted pathogenic genetic variants and population frequencies.
Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal AssociationMechanistic Insights into the Pathogenesis of Polycystic Kidney Disease.
CellsOxy210 Inhibits Hepatic Expression of Senescence-Associated, Pro-Fibrotic, and Pro-Inflammatory Genes in Mice During Development of MASH and in Hepatocytes In Vitro.
CellsTargeting TRAF6 inhibits cystogenesis in autosomal dominant polycystic kidney disease.
Biochemical pharmacologyLysine Methyltransferases SMYD2 and SMYD3: Emerging Targets in Kidney Diseases.
Kidney diseases (Basel, Switzerland)Nutritional considerations for designing ketogenic dietary interventions for people with Autosomal Dominant Polycystic Kidney Disease.
Journal of nephrologyRisk factors for perinatal and neonatal mortality in cases with congenital anomalies of the kidney and urinary tract: a nested cohort study.
Pediatric nephrology (Berlin, Germany)Urinary renal epithelial cells can be used for NPHP1 phenotyping and a personalized therapeutic strategy.
Journal of cell scienceCentriolar protein PIBF1 is required for craniofacial and forebrain development.
Developmental biologyPulmonary cysts as a diagnostic indicator of Birt-Hogg-Dubé syndrome in patients with renal neoplasm.
Insights into imagingHyperosmotic stimuli activate polycystin proteins to aid in urine concentration.
JCI insightA novel gene therapy for ARPKD based on CFTR.
American journal of physiology. Gastrointestinal and liver physiologyADPKD Progression by Variant Type and Molecular Domain Among Participants in the HALT PKD Trial.
Clinical journal of the American Society of Nephrology : CJASNGenetic Characterization of Lithuanian Patients With Cystic Kidney.
Clinical geneticsImportance of family history and health checkup for school-aged children for type IV collagen-associated nephropathy in hereditary kidney disease.
Journal of nephrologyIntegration of Genetics Into the Design and Conduct of Clinical Trials in Nephrology.
Seminars in nephrologyJoubert Syndrome in Children-A Comprehensive Analysis of Quality of Life, Functional Independence and Family Impact.
American journal of medical genetics. Part AStructure of CFTR bound to (R)-BPO-27 unveils a pore-blockage mechanism.
Nature communicationsA Diagnosis of Maturity-onset Diabetes of the Young Type 5 Provides Clarity and Broadens Reproductive Options.
JCEM case reportsCiliopathy: Senior-Løken Syndrome.
Advances in experimental medicine and biologyLeber Congenital Amaurosis.
Advances in experimental medicine and biologyTubulocystic Renal Cell Carcinoma With Pure Morphology and Confirmed "Wild Type" FH/2SC Immunophenotype: Clinicopathologic Series of 30 Patients.
The American journal of surgical pathologyTherapies in autosomal dominant polycystic kidney disease: beyond tolvaptan.
Current opinion in nephrology and hypertensionClinical and Genetic Characteristics of Senior-Loken Syndrome Patients in Korea.
GenesAutosomal Dominant Polycystic Kidney Disease: From Pathogenesis to Organoid Disease Models.
BiomedicinesThe nephronophthisis protein GLIS2/NPHP7 is required for the DNA damage response in kidney tubular epithelial cells.
American journal of physiology. Renal physiologyTargeting the endocannabinoid system to suppress mTORC1 hyperactivation in TSC-associated kidney disease.
American journal of physiology. Renal physiologyThe relevance of primary cilia in neurological disorders.
The Lancet. NeurologyNSD2 promotes cell durotaxis and drives the transition from polycystic kidney disease to tubulocystic renal cell carcinoma through integrin/FAK/AKT signaling.
OncogeneKidney Cysts in Autosomal Dominant Polycystic Kidney Disease and Alport Syndrome: Two Familial Cases Illustrating Diagnostic Challenges.
American journal of kidney diseases : the official journal of the National Kidney FoundationEmpowering women at the heart of autosomal dominant polycystic kidney disease: Addressing unique challenges gender-sensitive approach.
Women's health (London, England)Developing serum proteomics based prediction models of disease progression in ADPKD.
Nature communicationsClinic Examination and Gene Diagnosis for a Birt-Hogg-Dubé Syndrome Family With a Novel flcn Frameshift Mutation Causing Nonsense-Mediated mRNA Degradation.
Human mutationCharacterization of Monogenic Kidney Disease in Older Patients With CKD.
Kidney international reportsGenetic diagnosis and prenatal diagnosis of patients with cystic kidney disease in Southwest China.
BMC nephrologyCo-occurrence of Peutz-Jeghers syndrome and unilateral multicystic dysplastic kidney: a case report.
BMC nephrologyTargeting of a Bile Acid Receptor, Takeda G Protein Receptor 5 [TGR5], by a Novel First-in-Class Competitive Antagonist, SBI-364, Diminishes Hepatic and Renal Cystogenesis in Polycystic Liver and Kidney Disease.
ACS pharmacology & translational scienceBirt-Hogg-Dubé Syndrome-A Rare Cause of Recurrent Pneumothoraces in a Midshipman.
Military medicineValosin-containing protein in ciliary morphology: a novel target in ADPKD.
American journal of physiology. Renal physiologyChallenges in the Diagnosis and Care of Tuberous Sclerosis Complex in Resource-Limited Settings: A Case Report from Tanzania.
Case reports in neurologyUsing Large Genomic Biobanks to Generate Insights into Genetic Kidney Disease.
Seminars in nephrologyPatients With Mild ADPKD by Kidney Imaging but Low Estimated GFR.
Kidney international reportsDifferences in gut microbiome between autosomal dominant polycystic kidney disease with and without intracranial aneurysms.
Scientific reports[Identification of a novel deep intronic variant associated with Joubert syndrome through combined whole-genome sequencing and RNA sequencing].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Doença renal cística genética.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Doença renal cística genética
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Reversible cystogenesis in juvenile primate ADPKD models: evidence from PKD1 heterozygous monkeys.
- Heterogeneous regulation of fibroblast growth factor 23 in acute kidney injury, chronic kidney disease, and polycystic kidney disease: mechanisms, diagnostic utility, and clinical implications.
- Emodin retards renal cyst progression in ADPKD by inhibiting cell proliferation and decreasing oxidative stress.
- An NGS-based investigation of copy number variants in the diagnosis and severity of adult polycystic kidney disease.
- Epithelial Dynamics of Cystogenesis in Genetic Models of Autosomal Dominant Polycystic Kidney Disease.
- Cystic renal diseases: role of ultrasound. Part II, genetic cystic renal diseases.
- Cystic renal diseases: role of ultrasound. Part I, non-genetic cystic renal diseases.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:93587(Orphanet)
- MONDO:0019741(MONDO)
- GARD:19228(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55788849(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
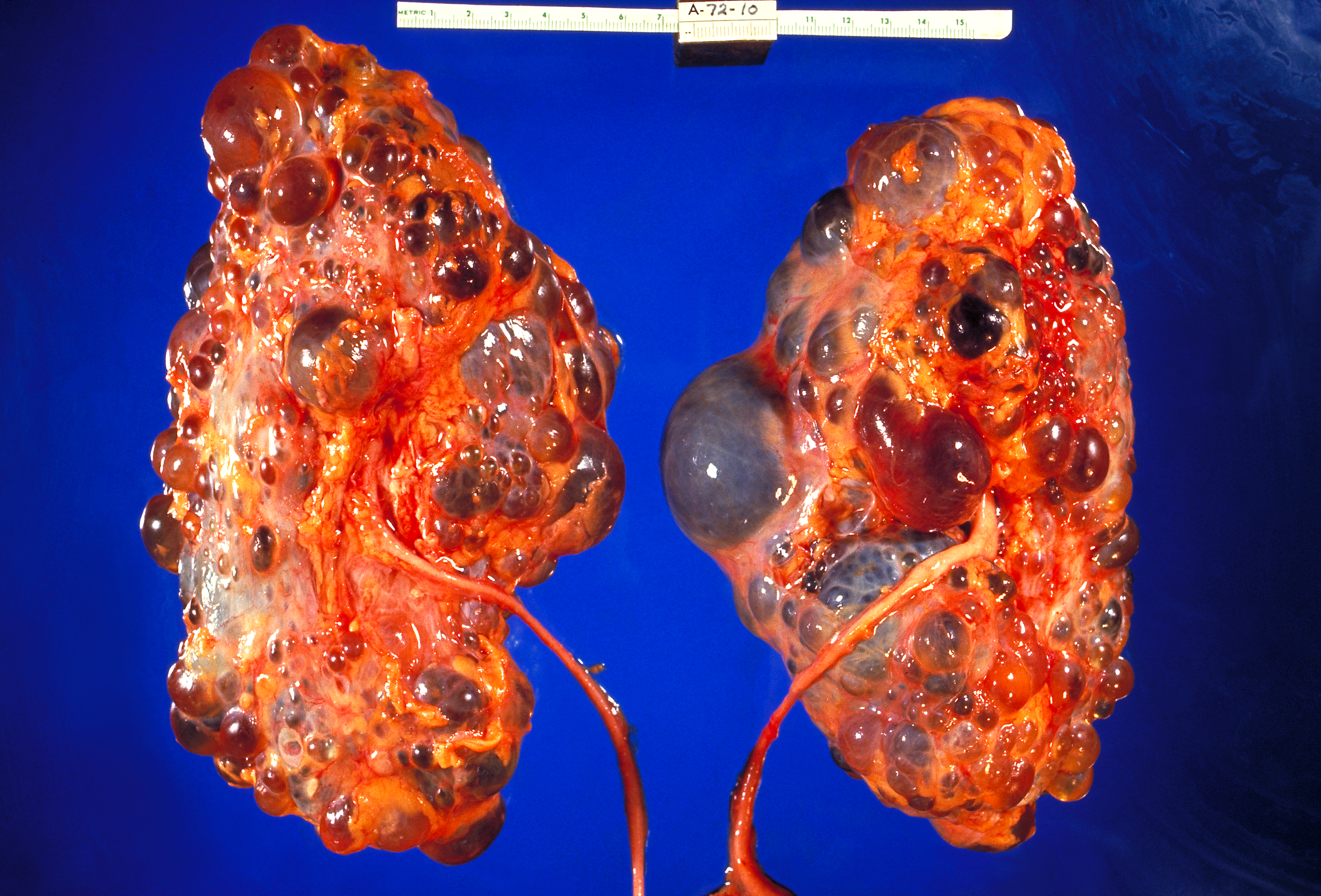
Doença renal cística genética
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos (literatura)
- fonte: Orphanet