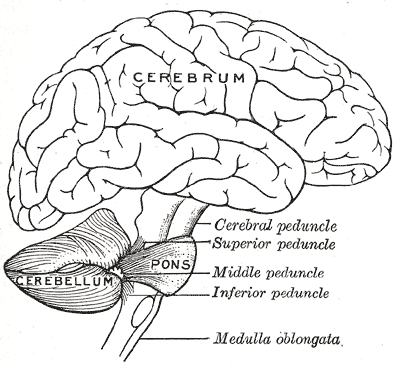
A hipoplasia pontocerebelar tipo 1 (PCH1), também conhecida como doença de Norman, é um grupo clínica e geneticamente heterogêneo de doenças autossômicas recessivas com início pré-natal caracterizada por atrofia muscular difusa secundária à hipoplasia pontocerebelar e degeneração das células do corno anterior da medula espinhal, resultando em morte precoce.
Introdução
O que você precisa saber de cara
A hipoplasia pontocerebelar tipo 1 (PCH1), também conhecida como doença de Norman, é um grupo clínica e geneticamente heterogêneo de doenças autossômicas recessivas com início pré-natal caracterizada por atrofia muscular difusa secundária à hipoplasia pontocerebelar e degeneração das células do corno anterior da medula espinhal, resultando em morte precoce.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 25 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 75 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
6 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Non-catalytic component of the RNA exosome complex which has 3'->5' exoribonuclease activity and participates in a multitude of cellular RNA processing and degradation events. In the nucleus, the RNA exosome complex is involved in proper maturation of stable RNA species such as rRNA, snRNA and snoRNA, in the elimination of RNA processing by-products and non-coding 'pervasive' transcripts, such as antisense RNA species and promoter-upstream transcripts (PROMPTs), and of mRNAs with processing defe
CytoplasmNucleusNucleus, nucleolus
Pontocerebellar hypoplasia 1C
A severe autosomal recessive neurodegenerative disease characterized by cerebellar and corpus callosum hypoplasia, abnormal myelination of the central nervous system, and spinal motor neuron disease. Affected individuals manifest failure to thrive, severe muscle weakness, spasticity and psychomotor retardation. Vision and hearing are impaired.
Transmembrane protein of the mitochondrial outer membrane that controls mitochondrial organization (PubMed:26168012, PubMed:27390132, PubMed:27543974). May regulate the assembly of the MICOS (mitochondrial contact site and cristae organizing system) complex which is essential to the biogenesis and dynamics of mitochondrial cristae, the inwards folds of the inner mitochondrial membrane (PubMed:27390132). Through its interaction with the EMC (endoplasmic reticulum membrane protein complex), could
Mitochondrion outer membrane
Neuropathy, hereditary motor and sensory, 6B, with optic atrophy
An autosomal recessive neurologic disorder characterized by early-onset optic atrophy, progressive visual loss, and peripheral sensorimotor neuropathy manifesting as axonal Charcot-Marie-Tooth disease, with variable age at onset and severity. Charcot-Marie-Tooth disease is a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. It is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies and primary peripheral axonal neuropathies. Peripheral axonal neuropathies are characterized by signs of axonal regeneration in the absence of obvious myelin alterations, and normal or slightly reduced nerve conduction velocities.
Non-catalytic component of the RNA exosome complex which has 3'->5' exoribonuclease activity and participates in a multitude of cellular RNA processing and degradation events. In the nucleus, the RNA exosome complex is involved in proper maturation of stable RNA species such as rRNA, snRNA and snoRNA, in the elimination of RNA processing by-products and non-coding 'pervasive' transcripts, such as antisense RNA species and promoter-upstream transcripts (PROMPTs), and of mRNAs with processing defe
CytoplasmNucleus, nucleolusNucleus
Pontocerebellar hypoplasia 1B
A severe autosomal recessive neurologic disorder characterized by a combination of cerebellar and spinal motor neuron degeneration beginning at birth. There is diffuse muscle weakness, progressive microcephaly, global developmental delay, and brainstem involvement.
Serine/threonine kinase involved in the regulation of key cellular processes including the cell cycle, nuclear condensation, transcription regulation, and DNA damage response (PubMed:14645249, PubMed:18617507, PubMed:19103756, PubMed:33076429). Controls chromatin organization and remodeling by mediating phosphorylation of histone H3 on 'Thr-4' and histone H2AX (H2aXT4ph) (PubMed:31527692, PubMed:37179361). It also phosphorylates KAT5 in response to DNA damage, promoting KAT5 association with chr
NucleusCytoplasmNucleus, Cajal body
Pontocerebellar hypoplasia 1A
A form of pontocerebellar hypoplasia, a disorder characterized by structural defects of the pons and cerebellum, evident upon brain imaging. PCH1A is an autosomal recessive form characterized by an abnormally small cerebellum and brainstem, central and peripheral motor dysfunction from birth, gliosis and spinal cord anterior horn cells degeneration resembling infantile spinal muscular atrophy. Additional features include muscle hypotonia, congenital contractures and respiratory insufficiency that is evident at birth.
Metallocarboxypeptidase that mediates protein deglutamylation of tubulin and non-tubulin target proteins (PubMed:22170066, PubMed:24022482, PubMed:30420557). Catalyzes the removal of polyglutamate side chains present on the gamma-carboxyl group of glutamate residues within the C-terminal tail of alpha- and beta-tubulin (PubMed:22170066, PubMed:24022482, PubMed:30420557). Specifically cleaves tubulin long-side-chains, while it is not able to remove the branching point glutamate (PubMed:24022482).
CytoplasmCytoplasm, cytosolNucleusMitochondrion
Neurodegeneration, childhood-onset, with cerebellar atrophy
An autosomal recessive disorder characterized by early onset of progressive neurodegeneration affecting the central and peripheral nervous systems. Clinical features include global developmental delay, impaired intellectual development, poor or absent speech, and motor abnormalities. Brain imaging shows cerebellar atrophy. Death in childhood may occur.
Non-catalytic component of the RNA exosome complex which has 3'->5' exoribonuclease activity and participates in a multitude of cellular RNA processing and degradation events. In the nucleus, the RNA exosome complex is involved in proper maturation of stable RNA species such as rRNA, snRNA and snoRNA, in the elimination of RNA processing by-products and non-coding 'pervasive' transcripts, such as antisense RNA species and promoter-upstream transcripts (PROMPTs), and of mRNAs with processing defe
CytoplasmNucleusNucleus, nucleolusNucleus, nucleoplasm
Pontocerebellar hypoplasia 1D
An autosomal recessive neurologic disorder with onset at birth or in infancy, and characterized by progressive axonal motor neuronopathy, severe generalized hypotonia, respiratory insufficiency, and cerebellar atrophy. Death in childhood may occur.
Variantes genéticas (ClinVar)
294 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 34 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
10 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Hipoplasia pontocerebelar tipo 1
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
[Pontocerebellar hypoplasia type 1F associated with homozygous variation of EXOSC1 gene in a neonate].
患儿,男,31分钟龄。胎儿期表现为重度生长受限,出生后表现为宫内发育迟缓(出生体重、头围、身长均低于同胎龄、同性别第3百分位)、小头畸形、鼻梁低、眼距宽、肌张力低下,可见全身花斑纹改变。实验室检查肾功能异常;头颅磁共振成像提示脑桥小脑发育不全、脑萎缩和髓鞘形成延迟;全外显子组测序发现EXOSC1基因c.547C>T(p.Arg183Trp)纯合变异。结合临床表现和基因检测,患儿诊断为EXOSC1基因纯合变异相关脑桥小脑发育不全1F型。.
Pontocerebellar Hypoplasia Type 1 and Associated Neuronopathies.
Pontocerebellar hypoplasia is a rare neurodegenerative syndrome characterized by severe hypoplasia or atrophy of pons and cerebellum that may be associated with other brain malformations, microcephaly, optic nerve atrophy, dystonia, ataxia and neuromuscular disorders. At this time, there are 17 variants of PCH distinguished by clinical presentation and distinctive radiological and biochemical features in addition to pontine and cerebellar hypoplasia. PCH1 is defined as PCH variant associated with anterior horn degeneration in the spinal cord with muscle weakness and hypotonia, and is associated with recessive variants in genes VRK1, EXOSC3, EXOSC8, EXOSC9 and SLC25A46. Neuromuscular manifestations may clinically present as amyotrophic lateral sclerosis (ALS), motor neuropathy (HMN) or neuronopathy (non-5q spinal muscular atrophy; SMA) or sensorimotor polyneuropathy (HMSN). Physiologic functions of PCH1-associated genes include regulation of RNA metabolism, mitochondrial fission and neuronal migration. Overall, complex phenotypes associated with PCH1 gene variants ranging from PCH and related neurodevelopmental disorders combined with neuromuscular disorders to isolated neuromuscular disorders have variable outcomes with isolated neuromuscular disorders typically having later onset with better outcomes. Improved understanding of pathogenesis of pontocerebellar hypoplasia and its association with motor neuronopathies and peripheral neuropathies may provide us with valuable insights and lead to potential new therapeutic targets for neurodegenerative disorders.
The phenotyping dilemma in VRK1-related motor neuron disease: a Turkish family with young-onset amyotrophic lateral sclerosis caused by a novel mutation.
Objective: Vaccinia-related kinase 1 (VRK1)-related disease is an extremely rare autosomal recessive disorder primarily affecting the peripheral and/or central nervous system. In this report, we describe the genetic and clinical features of two siblings from a Turkish family presenting with an amyotrophic lateral sclerosis (ALS) phenotype due to a novel homozygous VRK1 mutation, and discuss the broad phenotypic spectrum associated with pathogenic variants in this gene. Methods: We analyzed the demographic data, clinical histories, neurological examinations, laboratory findings, and genetic results of 53 patients, including our cases, derived from 27 different reports. Results: Whole-exome sequencing identified a novel homozygous missense mutation, c.700A > G (p.Asn234Asp), in the VRK1 gene in two affected siblings. The characteristic features of the ALS phenotype included a recessive inheritance pattern, motor deficits with onset in the lower limbs, pyramidal tract signs, and a muscle magnetic resonance imaging (MRI) pattern demonstrating preferential involvement of the posterior compartments of the leg and thigh. The most common phenotypes associated with VRK1 mutations were ALS (18/53, 34%) and distal hereditary motor neuropathy (dHMN) (14/53, 26.4%), followed by pontocerebellar hypoplasia type 1 (7/53, 13.2%), hereditary motor and sensory neuropathy (5/53, 9.4%), autosomal recessive primary microcephaly with brain malformations (4/53, 7.5%), and spastic paraplegia (2/53, 3.8%). The ALS phenotype exhibited a significantly earlier mean age of onset compared to the dHMN phenotype (p = 0.015; 15.3 ± 11.5 and 27 ± 15.5 years, respectively). Conclusion: Our findings highlight the importance of investigating VRK1 mutations in patients with young-onset familial ALS. Furthermore, this report provides a systematic classification of the phenotype definitions associated with VRK1 mutations.
Pontocerebellar hypoplasia associated with p.Arg183Trp homozygous variant in EXOSC1 gene: A case report.
Pontocerebellar hypoplasia (PCH) is a heterogeneous group of rare neurodegenerative disorders characterized by a wide phenotypic range including severe motor and cognitive impairments, microcephaly, distinctive facial features, and other features according to the type. Several classes of PCH1 have been linked to mutations in the evolutionarily conserved RNA exosome complex that consists of nine subunits (EXOSC1 to EXOSC9) and facilitates the degradation and processing of cytoplasmic and nuclear RNA from the 3' end. Only a single individual with an EXOSC1 mutation was reported with clinical features of PCH type 1 (PCH1F). Here, we report a 3-month-old female with PCH and additional clinical features not previously reported to be associated with PCH1, including dilated cardiomyopathy. On assessment, failure to thrive, microcephaly, distinctive facial features, and bluish sclera, were noted. Whole-exome sequencing was performed and revealed a novel homozygous missense variant c.547C > T (p.Arg183Trp) in the EXOSC1 gene. Functional studies in a budding yeast model that expresses the human EXOSC1 variant Arg183Trp show a slow-growth phenotype, whereas the previously identified PCH1F allele EXOSC1-Ser35Leu is lethal, indicating impaired exosome function for both of these variants. The protein levels of both EXOSC1 variants are reduced compared with wild-type when expressed in budding yeast. Herein, we ascertain the second case of PCH associated with a EXOSC1 variant that causes defects in RNA exosome function and provide a model organism system to distinguish between benign and pathogenic variants in EXOSC1.
Molecularly confirmed pontocerebellar hypoplasia in a large family from Slovakia with four severely affected children.
Pontocerebellar hypoplasia type 1 (PCH1) is characterized by a central and peripheral motor dysfunction associated with anterior horn cell degeneration, similar to spinal muscular atrophy (SMA). We analysed three probands (later discovered to be siblings) suspected to have severe SMA, however, not confirmed by genetic test. Clinical-exome analysis (Illumina) was performed to identify causative variants, followed by Sanger sequencing confirmation in probands and other 10 family members. Homozygous pathogenic variant c.92G>C (p.(Gly31Ala)) in the Exosome Component 3 (EXOSC3) gene was found in all 3 probands, thus confirming the diagnosis of a severe form of PCH1B. The parents and six siblings were carriers, while one sibling was homozygous for a reference allele. This variant is frequent in the Czech Roma population, where it is considered a founder mutation. Haplotype analysis in this largest reported PCH1B family showed that our patients inherited from their father (of Roma origin) a haplotype identical to that found in the Czech Roma population, thus indicating these alleles have a common origin. This EXOSC3 variant is rare among the general population but most likely frequent also among Roma people in Slovakia. PCH1B should be considered for a differential diagnosis in infants manifesting SMA-like phenotype, especially if of Roma origin (Tab. 1, Fig. 1, Ref. 22). Text in PDF www.elis.sk Keywords: pontocerebellar hypoplasia, PCH1B, EXOSC3, SMA plus syndromes, pathogenic sequence variant.
Publicações recentes
Pontocerebellar Hypoplasia Type 1 and Associated Neuronopathies.
The phenotyping dilemma in VRK1-related motor neuron disease: a Turkish family with young-onset amyotrophic lateral sclerosis caused by a novel mutation.
Pontocerebellar hypoplasia associated with p.Arg183Trp homozygous variant in EXOSC1 gene: A case report.
Molecularly confirmed pontocerebellar hypoplasia in a large family from Slovakia with four severely affected children.
Bi-allelic missense variant, p.Ser35Leu in EXOSC1 is associated with pontocerebellar hypoplasia.
📚 EuropePMC251 artigos no totalmostrando 18
Pontocerebellar Hypoplasia Type 1 and Associated Neuronopathies.
GenesThe phenotyping dilemma in VRK1-related motor neuron disease: a Turkish family with young-onset amyotrophic lateral sclerosis caused by a novel mutation.
Amyotrophic lateral sclerosis & frontotemporal degeneration[Pontocerebellar hypoplasia type 1F associated with homozygous variation of EXOSC1 gene in a neonate].
Zhonghua er ke za zhi = Chinese journal of pediatricsPontocerebellar hypoplasia associated with p.Arg183Trp homozygous variant in EXOSC1 gene: A case report.
American journal of medical genetics. Part AMolecularly confirmed pontocerebellar hypoplasia in a large family from Slovakia with four severely affected children.
Bratislavske lekarske listyNew subtype of PCH1C caused by novel EXOSC8 variants in a 16-year-old Spanish patient.
Neuromuscular disorders : NMDBi-allelic missense variant, p.Ser35Leu in EXOSC1 is associated with pontocerebellar hypoplasia.
Clinical geneticsNovel EXOSC9 variants cause pontocerebellar hypoplasia type 1D with spinal motor neuronopathy and cerebellar atrophy.
Journal of human geneticsBiallelic variants in AGTPBP1, involved in tubulin deglutamylation, are associated with cerebellar degeneration and motor neuropathy.
European journal of human genetics : EJHGExpanded PCH1D phenotype linked to EXOSC9 mutation.
European journal of medical geneticsDe novo variant in KIF26B is associated with pontocerebellar hypoplasia with infantile spinal muscular atrophy.
American journal of medical genetics. Part AA Chemical Biology Approach to Model Pontocerebellar Hypoplasia Type 1B (PCH1B).
ACS chemical biologySarcomeric disorganization and nemaline bodies in muscle biopsies of patients with EXOSC3-related type 1 pontocerebellar hypoplasia.
Muscle & nervePontocerebellar hypoplasia type 1 for the neuropediatrician: Genotype-phenotype correlations and diagnostic guidelines based on new cases and overview of the literature.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyWhole exome sequencing diagnoses the first fetal case of Bainbridge-Ropers syndrome presenting as pontocerebellar hypoplasia type 1.
Birth defects researchRecessive mutation in EXOSC3 associates with mitochondrial dysfunction and pontocerebellar hypoplasia.
MitochondrionExtension of the phenotype of biallelic loss-of-function mutations in SLC25A46 to the severe form of pontocerebellar hypoplasia type I.
Clinical genetics[Pontocerebellar hypoplasia is a rare cause of floppy infant syndrome].
Ugeskrift for laegerAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Hipoplasia pontocerebelar tipo 1.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Hipoplasia pontocerebelar tipo 1
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- [Pontocerebellar hypoplasia type 1F associated with homozygous variation of EXOSC1 gene in a neonate].
- Pontocerebellar Hypoplasia Type 1 and Associated Neuronopathies.
- The phenotyping dilemma in VRK1-related motor neuron disease: a Turkish family with young-onset amyotrophic lateral sclerosis caused by a novel mutation.
- Pontocerebellar hypoplasia associated with p.Arg183Trp homozygous variant in EXOSC1 gene: A case report.
- Molecularly confirmed pontocerebellar hypoplasia in a large family from Slovakia with four severely affected children.
- Bi-allelic missense variant, p.Ser35Leu in EXOSC1 is associated with pontocerebellar hypoplasia.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2254(Orphanet)
- MONDO:0016396(MONDO)
- GARD:10704(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q18966144(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Hipoplasia pontocerebelar tipo 1
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata