A síndrome da contratura congênita letal tipo 2 é uma síndrome rara de artrogripose caracterizada por múltiplas contatoras congênitas (tipicamente cotovelos estendidos e joelhos flexionados), micrognatia, degeneração das células do corno anterior, atrofia da musculatura esquelética (principalmente nos membros inferiores), presença de bexiga urinária acentuadamente distendida e ausência de hidropisia, pterígio e fraturas ósseas. Outras anomalias craniofaciais (por exemplo, fenda palatina, paralisia facial) e oculares (por exemplo, anisocoria, descolamento de retina) podem ser observadas adicionalmente. A doença é geralmente letal para o recém-nascido, no entanto, foi relatada sobrevivência até a adolescência.
Introdução
O que você precisa saber de cara
A síndrome da contratura congênita letal tipo 2 é uma síndrome rara de artrogripose caracterizada por múltiplas contatoras congênitas (tipicamente cotovelos estendidos e joelhos flexionados), micrognatia, degeneração das células do corno anterior, atrofia da musculatura esquelética (principalmente nos membros inferiores), presença de bexiga urinária acentuadamente distendida e ausência de hidropisia, pterígio e fraturas ósseas. Outras anomalias craniofaciais (por exemplo, fenda palatina, paralisia facial) e oculares (por exemplo, anisocoria, descolamento de retina) podem ser observadas adicionalmente. A doença é geralmente letal para o recém-nascido, no entanto, foi relatada sobrevivência até a adolescência.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 5 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 14 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisTyrosine-protein kinase that plays an essential role as cell surface receptor for neuregulins. Binds to neuregulin-1 (NRG1) and is activated by it; ligand-binding increases phosphorylation on tyrosine residues and promotes its association with the p85 subunit of phosphatidylinositol 3-kinase (PubMed:20682778). May also be activated by CSPG5 (PubMed:15358134). Involved in the regulation of myeloid cell differentiation (PubMed:27416908)
Cell membraneSecreted
Lethal congenital contracture syndrome 2
A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology.
Variantes genéticas (ClinVar)
39 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 219 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
15 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de contratura congênita letal tipo 2
Centros de Referência SUS
24 centros habilitados pelo SUS para Síndrome de contratura congênita letal tipo 2
Centros para Síndrome de contratura congênita letal tipo 2
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Deciphering DST-associated disorders: biallelic variants affecting DST-b cause a congenital myopathy.
The dystonin gene (DST) encodes three major isoforms, DST-a, DST-b and DST-e. Biallelic pathogenic variants in DST have previously been associated with two allelic monogenic disorders: hereditary sensory and autonomic neuropathy type VI (caused by a loss of DST-a) and epidermolysis bullosa simplex 3 (caused by a loss of DST-e). We investigated patients diagnosed with congenital myopathy using exome or genome sequencing. In 19 affected individuals from 14 unrelated families, we identified nine different variants in biallelic state located in exons 40-41, specific to DST-b. Affected individuals presented with severe neonatal myopathy characterized by arthrogryposis, hypotonia and dilated cardiomyopathy. Postnatal CPAP ventilation was required in nine patients, and seven died within the first three years of life. Survivors showed an improvement of symptoms, with the oldest three patients, now over 25 years old, exhibiting normal cognition and being ambulatory. RNA analyses demonstrated that transcripts encoding DST-b are predominantly expressed in skeletal muscle, heart tissue and cultured fibroblasts, but not in brain, matching the phenotypic spectrum. Patient-derived fibroblasts exhibited reduced DST mRNA expression. Proteomic analysis confirmed a reduction of DST protein levels due to an absence of the DST-b isoform. Muscle biopsies from four patients aged 1 month to 3 years revealed mild, non-specific myopathic changes. Ultrastructural analysis in three individuals showed mild and focal myofibrillar disruption and non-specific undulating nuclear membranes, with these changes observed in two cases each. Additionally, we identified two homozygous variants affecting both DST-a and DST-b isoforms in four patients from two unrelated families; all presented with severe arthrogryposis and died intrauterine or shortly after birth. Genotype-phenotype correlation in these patients and previously published cases with respective variants resulted in the definition of a DST-associated lethal congenital contracture syndrome. Our findings demonstrate that biallelic variants exclusively affecting DST-b cause an autosomal recessive congenital myopathy. Variants that also impact DST-a besides DST-b result in a more severe, lethal congenital contracture syndrome. The location of the variant within DST allows for phenotype prediction. We propose redefining DST as a disease-associated gene linked to four distinct allelic disease phenotypes.
Variants in DOK7 results in fetal akinesia deformation sequence: A case report and review of literature.
We report the third case of FADS due to biallelic DOK7 variants, which further strengthens the association of DOK7 with this lethal phenotype and lack of genotype phenotype correlation.
Lethal respiratory course and additional features expand the phenotypic spectrum of PIEZO2-related distal arthrogryposis type 5.
Distal arthrogryposes (DA) are a group of conditions presenting with multiple congenital contractures in the distal joints. The 10 types of DA are distinguished by different extra-articular manifestations. Heterozygous gain-of-function variants in PIEZO2 are known to cause a spectrum of DA conditions including DA type 3, DA type 5, and possibly Marden Walker syndrome, which are usually distinguished by the presence of cleft palate (DA3), ptosis and restriction in eye movements (DA5), and specific facial abnormalities and central nervous system involvement, respectively. We report on a boy with a recurrent de novo heterozygous PIEZO2 variant in exon 20 (NM_022068.3: c.2994G > A, p.(Met998Ile); NM_001378183.1: c.3069G > A, p.(Met1023Ile)), who presented at birth with DA and later developed respiratory insufficiency. His phenotype broadly fits the PIEZO2 phenotypic spectrum and potentially extends it with novel phenotypic features of pretibial linear vertical crease, immobile skin, immobile tongue, and lipid myopathy.
Involvement of muscle satellite cell dysfunction in neuromuscular disorders: Expanding the portfolio of satellite cell-opathies.
Neuromuscular disorders are a heterogeneous group of acquired or hereditary conditions that affect striated muscle function. The resulting decrease in muscle strength and motility irreversibly impacts quality of life. In addition to directly affecting skeletal muscle, pathogenesis can also arise from dysfunctional crosstalk between nerves and muscles, and may include cardiac impairment. Muscular weakness is often progressive and paralleled by continuous decline in the ability of skeletal muscle to functionally adapt and regenerate. Normally, the skeletal muscle resident stem cells, named satellite cells, ensure tissue homeostasis by providing myoblasts for growth, maintenance, repair and regeneration. We recently defined 'Satellite Cell-opathies' as those inherited neuromuscular conditions presenting satellite cell dysfunction in muscular dystrophies and myopathies (doi:10.1016/j.yexcr.2021.112906). Here, we expand the portfolio of Satellite Cell-opathies by evaluating the potential impairment of satellite cell function across all 16 categories of neuromuscular disorders, including those with mainly neurogenic and cardiac involvement. We explore the expression dynamics of myopathogenes, genes whose mutation leads to skeletal muscle pathogenesis, using transcriptomic analysis. This revealed that 45% of myopathogenes are differentially expressed during early satellite cell activation (0 - 5 hours). Of these 271 myopathogenes, 83 respond to Pax7, a master regulator of satellite cells. Our analysis suggests possible perturbation of satellite cell function in many neuromuscular disorders across all categories, including those where skeletal muscle pathology is not predominant. This characterisation further aids understanding of pathomechanisms and informs on development of prognostic and diagnostic tools, and ultimately, new therapeutics.
[Analysis of CNTNAP1 gene variants in a Chinese pedigree affected with lethal congenital contracture syndrome type 7].
To explore the genetic basis for a couple who had developed polyhydramnios during three pregnancies and given birth to two liveborns featuring limb contracture, dyspnea and neonatal death. Whole-exome sequencing (WES) was carried out on fetal tissue and peripheral blood samples from the couple. Suspected variants were verified by Sanger sequencing. The fetus was found to harbor homozygous nonsense c.3718C>T (p.Arg1240Ter) variants of the CNTNAP1 gene, which were respectively inherited from its mother and father. The variant was unreported previously. According to the guidelines of the American College of Medical Genetics and Genomics, the variant was predicted to be pathogenic (PVS1+PM2+PP4). The novel homozygous nonsense variants of the CNTNAP1 gene probably underlay the lethal congenital contracture syndrome type 7 (LCCS7) in this pedigree. Above finding has enabled genetic counseling and prenatal diagnosis for the family.
Publicações recentes
A case of CNTNAP1 gene-related abnormality and literature review.
Expanding the Prenatal Phenotype of Lethal Congenital Contracture Syndrome 11: Novel Homozygous GLDN Variant in a Family With Recurrent Affected Fetuses.
Human CNTNAP1 Variants Associated With Severe Neurological Deficits: Additional Cases and Literature Review.
ADGRG6-related disorder: a novel mutation resulting in distal arthrogryposis and a patchy neuropathy.
In vivo modeling of lethal congenital contracture syndrome 1 suggests pathomechanisms in cellular stress responses.
📚 EuropePMC16 artigos no totalmostrando 10
Deciphering DST-associated disorders: biallelic variants affecting DST-b cause a congenital myopathy.
Brain : a journal of neurologyVariants in DOK7 results in fetal akinesia deformation sequence: A case report and review of literature.
Clinical geneticsLethal respiratory course and additional features expand the phenotypic spectrum of PIEZO2-related distal arthrogryposis type 5.
American journal of medical genetics. Part AInvolvement of muscle satellite cell dysfunction in neuromuscular disorders: Expanding the portfolio of satellite cell-opathies.
European journal of translational myology[Analysis of CNTNAP1 gene variants in a Chinese pedigree affected with lethal congenital contracture syndrome type 7].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsCNTNAP1 Mutations and Their Clinical Presentations: New Case Report and Systematic Review.
Case reports in medicinePremature aging syndromes: From patients to mechanism.
Journal of dermatological scienceExpanding the Clinical Spectrum of Phenotypes Caused by Pathogenic Variants in PLOD2.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchPhenotype variability and histopathological findings in patients with a novel DNM2 mutation.
Neuropathology : official journal of the Japanese Society of NeuropathologyA recurrent germline mutation in the PIGA gene causes Simpson-Golabi-Behmel syndrome type 2.
American journal of medical genetics. Part AAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de contratura congênita letal tipo 2.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de contratura congênita letal tipo 2
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Deciphering DST-associated disorders: biallelic variants affecting DST-b cause a congenital myopathy.
- Variants in DOK7 results in fetal akinesia deformation sequence: A case report and review of literature.
- Lethal respiratory course and additional features expand the phenotypic spectrum of PIEZO2-related distal arthrogryposis type 5.
- Involvement of muscle satellite cell dysfunction in neuromuscular disorders: Expanding the portfolio of satellite cell-opathies.
- [Analysis of CNTNAP1 gene variants in a Chinese pedigree affected with lethal congenital contracture syndrome type 7].Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics· 2022· PMID 35076918mais citado
- A case of CNTNAP1 gene-related abnormality and literature review.
- Expanding the Prenatal Phenotype of Lethal Congenital Contracture Syndrome 11: Novel Homozygous GLDN Variant in a Family With Recurrent Affected Fetuses.
- Human CNTNAP1 Variants Associated With Severe Neurological Deficits: Additional Cases and Literature Review.
- ADGRG6-related disorder: a novel mutation resulting in distal arthrogryposis and a patchy neuropathy.
- In vivo modeling of lethal congenital contracture syndrome 1 suggests pathomechanisms in cellular stress responses.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:137776(Orphanet)
- OMIM OMIM:607598(OMIM)
- MONDO:0011868(MONDO)
- GARD:9177(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q26492778(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
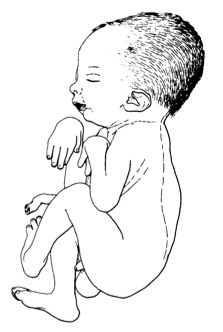
Síndrome de contratura congênita letal tipo 2
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata